Within the dermis is a fibrillar matrix, termed ground substance, composed of proteoglycans and glycosaminoglycans. These acid mucopolysaccharides, produced by fibroblasts, are highly hygroscopic, binding about 1000 times their own volume in water. They are critical in holding water in the dermis and are responsible for dermal volume and texture. Normally, the sulfated acid mucopolysaccharide chondroitin sulfate and heparin are the primary dermal mucins. In certain diseases, fibroblasts produce abnormally large amounts of acid mucopolysaccharides, usually hyaluronic acid. These acid mucopolysaccharides (mucin) accumulate in large amounts in the dermis and may be visible histopathologically as pale-blue, granular or amorphous material between collagen bundles. They are often not visualized with hematoxylin and eosin stains because the water they bind is removed in processing, so the presence of increased mucin is suspected by the presence of large, empty spaces between the collagen bundles. Acid mucopolysaccharides can be detected by special stains, such as colloidal iron, alcian blue, and toluidine blue. Incubation of the tissue with hyaluronidase eliminates the staining, confirming the presence of hyaluronic acid.
Increased dermal mucin may result from many diseases and is a normal component of wound healing. The mucinoses are diseases in which production of increased amounts of mucin is the primary process. Mucin may also accumulate in the skin as a secondary phenomenon in lupus erythematosus, dermatomyositis, Degos disease, granuloma annulare, and cutaneous tumors, or after therapies such as psoralen plus ultraviolet A (PUVA) or retinoids. The genetic diseases in which mucin accumulates as a result of inherited metabolic abnormalities are termed the mucopolysaccharidoses (see Chapter 26 ). Myxedema and pretibial myxedema are reviewed in Chapter 24 .
Lichen Myxedematosus
The terminology used to describe disorders in the lichen myxedematosus group has varied widely over the years; the 2001 classification of Rongioletti and Rebora is used here. A generalized form, scleromyxedema, is usually accompanied by a monoclonal gammopathy (lambda more commonly than kappa) and may have systemic organ involvement. Five localized forms are recognized, characterized by a lack of a monoclonal antibody and systemic disease. Also, patients may have disease that does not fit into these subsets, and their condition is termed atypical or intermediate in type. Mucin deposition secondary to thyroid disease is excluded from the classification.
Generalized Lichen Myxedematosus
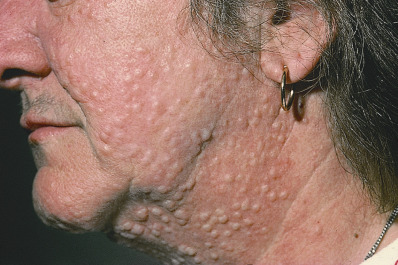
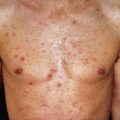
Scleromyxedema affects both men and women and generally appears between ages 30 and 80. It is chronic and progressive. The primary lesions are multiple, waxy, 2–4 mm, dome-shaped or flat-topped papules ( Fig. 9.1 ). They may coalesce into plaques ( Fig. 9.2 ) or may be arranged in linear arrays. Less often, urticarial, nodular, or even annular lesions are seen. The dorsal hands, face, elbows, and extensor extremities are most frequently affected ( Fig. 9.3 ). Mucosal lesions are absent.
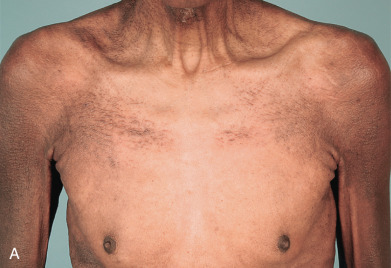
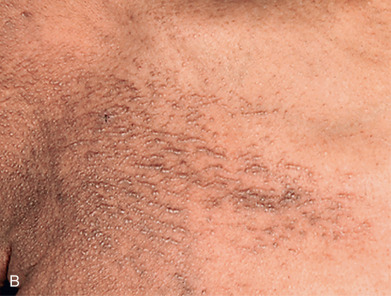
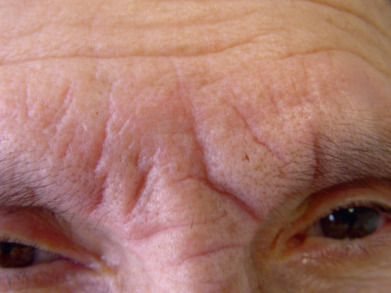
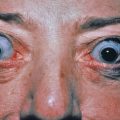
A diffuse infiltration develops, leading to “woody” sclerosis of the skin. A reduced range of motion of the mouth, hands, and extremities may follow ( Fig. 9.4 ). On the glabella and forehead, coalescence of lesions leads to the prominent furrowing of a “leonine facies.” At the proximal interphalangeal joint, induration surrounding a centrally depressed area has been called the “doughnut sign.” Pruritus may occur.
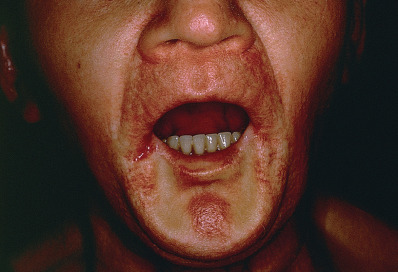
Scleromyxedema is often associated with visceral disease. Gastrointestinal findings are most common. Dysphagia from esophageal involvement often occurs, and the stomach or intestine may also be affected. Pulmonary complications with dyspnea caused by restrictive or obstructive disease are also common. Proximal muscle weakness with an inflammatory myopathy or a nonspecific vacuolar change may occur. Carpal tunnel syndrome occurs in 10% of patients. Arthralgia or inflammatory arthritis frequently develop. Disease-specific adenopathy and renal impairment may be present.
The most serious systemic findings are cardiac, hematologic, and neurologic manifestations. Peripheral neuropathies and central nervous system disturbances can occur, including confusion, dizziness, dysarthria, ascending paralysis, seizures, syncope, and coma. The latter conditions have been called “dermato-neuro syndrome” and may be due to disruption in perfusion by the excessive immunoglobulins leading to an encephalopathy. Middle-aged men are most frequently affected, and one third of these patients demonstrate recurrent symptoms. Plasmapheresis and intravenous immune globulin (IVIG) may result in dramatic recovery from this life-threatening emergency. Visceral disease can be fatal.
Criteria for inclusion in the scleromyxedema category include mucin deposition, fibroblast proliferation and fibrosis, normal thyroid function tests, and presence of a monoclonal gammopathy. Approximately 10% of patients do not have a gammopathy on initial evaluation. Association with hepatitis C, diabetes mellitus, and multiple sclerosis has also been reported. The gammopathy is usually an immunoglobulin G–λ (IgG-λ) type, suggesting an underlying plasma cell dyscrasia. Bone marrow examination may be normal or may reveal increased numbers of plasma cells or frank multiple myeloma.
Clinical and histologic features are usually diagnostic. Skin biopsies of early papular lesions demonstrate a proliferation of fibroblasts with mucin and many small collagen fibers. The papules generally appear more fibrotic than mucinous. Over time, fibroblast nuclei become less numerous, and collagen fibers become thickened.
Many clinical findings in scleromyxedema are also found in systemic scleroderma, including cutaneous sclerosis, Raynaud phenomenon, dysphagia, and carpal tunnel syndrome. This distinction in some cases may be difficult without a biopsy. Other infiltrative disorders, such as amyloidosis, must be excluded. Nephrogenic systemic fibrosis presents with skin thickening in the setting of renal failure and gadolinium exposure (see Chapter 8 ). In its earliest form, it includes mucin along with collagen deposition with a proliferation of CD34+ cells in the dermis. The histologic findings are identical to those of scleromyxedema, and a first report referred to a scleromyxedema-like disease associated with renal failure. The clinical findings are dominated by fibrosis (see Chapter 8 ).
Treatment of scleromyxedema is difficult and usually undertaken in concert with an oncologist. Therapy is targeted to treat the gammopathy (if present). Typical therapies may include immunosuppressive agents, especially melphalan, bortezomib, or cyclophosphamide, with or without plasma exchange and high-dose prednisone. IVIG has also been used with success. Thalidomide and the newer thalidomide analog lenalinomide have shown benefit in patients with or without a paraproteinemia and may provide a long-term maintenance option. Temporary remission of progressive visceral disease may occur. These short-term benefits must be weighed against the increase in malignancies and sepsis complicating such therapy. Chances of remission are enhanced by the use of autologous stem cell transplantation with high-dose melphalan.
Skin-directed therapy may also be used. Physical therapy is indicated. Retinoids, plasmapheresis, extracorporeal photochemotherapy, grenz ray and electron beam therapy, PUVA, thalidomide, interferon-α (IFN-α), cyclosporine, topical dimethyl sulfoxide, and topical and intralesional hyaluronidase and corticosteroids have all produced improvement in the skin of select patients. Many others, however, have not benefited, and visceral disease is usually not affected. Ultraviolet B (UVB) light and IFN-α have exacerbated scleromyxedema.
Occasional patients are reported who spontaneously remit even after many years of disease; however, scleromyxedema remains a therapeutic challenge, and the overall prognosis is poor.
Localized Lichen Myxedematosus
The localized variants of lichen myxedematosus lack visceral involvement or an associated gammopathy. As a group, they are benign but often persistent. No therapy is reliably effective in any of the localized forms of lichen myxedematosus. Because there is no gammopathy, visceral involvement, or associated thyroid disease in any of the variants, often no treatment is needed. Shave excision or carbon dioxide (CO 2 ) ablation are other options for individual lesions. Spontaneous resolution may occur in all varieties.
Discrete Papular Lichen Myxedematosus
Discrete papular lichen myxedematosus is characterized by the occurrence of waxy, 2–5 mm, firm, flesh-colored papules, usually confined to the limbs or trunk. The papules may have an erythematous or yellowish hue, may coalesce into nodules or plaques, and may number into the hundreds. Nodules may occasionally be the predominant lesion present, with few or absent papules. The underlying skin is not indurated, and there is no associated gammopathy or internal involvement. Biopsy reveals the presence of mucin in the upper and middle dermis. Fibroblast proliferation is variable, but collagen deposition is minimal. The slow accumulation of papules is the usual course, without the development of a gammopathy or internal manifestations. Occasional cases may spontaneously involute.
Patients with advanced human immunodeficiency virus (HIV) disease have been reported to develop mucinous papules, usually widespread, unassociated with a paraprotein. It is usually seen in patients diagnosed with acquired immunodeficiency syndrome (AIDS), in patients with multiple infectious complications. These lesions may occur in association with an eczematous dermatitis or on normal skin. If associated with an eczematous dermatitis, the lesions often clear if the eczema is controlled. Lesions on normal skin may respond to systemic retinoid therapy. Intralesional hyaluronidase has been used for lesions of papular mucinosis. At times, spontaneous remission occurs. The authors have also seen a patient with AIDS and true scleromyxedema with visceral involvement, and two patients have been reported with acral persistent papular mucinosis.
Acral Persistent Papular Mucinosis
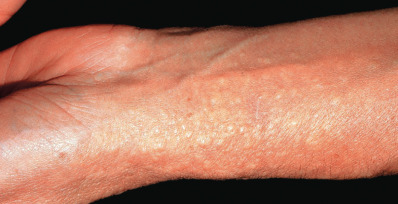
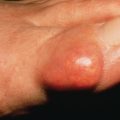
Patients with acral persistent papular mucinosis have a few to more than 100 bilaterally symmetric, 2–5 mm, flesh-colored papules localized to the hands and wrists ( Fig. 9.5 ). The knees, calves, or elbows may also be involved in a minority of patients. The face and trunk are spared. Women outnumber men by 5 : 1. The course is one of persistence and slow progression. Two involved sisters have been reported. Histologically, there is a collection of upper dermal mucin with minimal or no increase in fibroblasts. Electrocoagulation of these lesions was reported to result in no recurrence in 6 months and topical tacrolimus was also reported to have some benefit.
Self-Healing Papular Mucinosis
Self-healing papular mucinosis occurs in a juvenile and an adult form. The juvenile variant, also called self-healing juvenile cutaneous mucinosis, is a rare but distinct disorder characterized by the sudden onset of skin lesions and polyarthritis. Children, usually between ages 5 and 15, are affected. Familial cases are reported. Skin lesions are ivory-white papules of the head, neck, trunk, and typically the periarticular regions; deep nodules on the face and periarticular sites; and hard edema of the periorbital area and face. An acute arthritis affects the knees, elbows, and hand joints. In the adult form, papular lesions occur, usually without the associated joint symptoms ( Fig. 9.6 ). Histology of the skin lesions reveals dermal mucin with minimal fibroblastic proliferation or collagen deposition. Although the initial presentation is worrisome, the prognosis is excellent. Spontaneous resolution without sequelae occurs over several months.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


