Collagen
Many types of collagen have been identified in tissues of vertebrates ( Table 25.1 ). Collagens help form the support structure and scaffolding for many parts of the body, including tissues, blood vessels, and bones. Fibrillar collagens (types I, II, III, V, and XI) form fibrils that are among the most abundant proteins in the body. Type I collagen accounts for 60%–90% of the dry weight of skin, ligaments, and demineralized bone. Type III collagen is abundant in fetal skin and blood vessels. It comprises 35% of the collagen in normal adult skin, but up to 40% in inflamed skin in the setting of contact dermatitis. Basement membrane–associated collagen is made up of types IV and VII. Fiber-associated collagens (types VIII, IX, and XIV) are found on the surface of type I and II collagens and are believed to serve as flexible spacers among fibrils. Fibril-associated collagens with interrupted triple helices (FACITs) do not form fibrils themselves but are found attached to the surfaces of preexisting fibrils of the fibril-forming collagens. FACITs are composed of types IX, XII, XIV, XVI, XIX, XX, and XXI. Network-forming collagens are sheets formed from types VIII and X. Studies on types XV, XVII, and XIX demonstrate their widespread presence in basement membranes, particularly vascular endothelium, which may represent a new subgroup of collagens associated with angiogenic and pathologic processes. Type XVII collagen is also known as BP180, and contains the target antigens for several immunobullous diseases. Type VII collagen contains the target antigens for bullous lupus and epidermolysis bullosa acquisita. Type II collagen contains the target antigens for relapsing polychondritis. Genetic mutations leading to defects in or absence of various collagens (VII and XVII) can lead to epidermolysis bullosa.
| Collagen Type | Gene * | Chromosome | Tissue Distribution |
|---|---|---|---|
| I | COL1A1–2 | 17q21.3–q22 | Skin, bone, tendon |
| I-trimer | Tumors, cell cultures, skin, liver | ||
| II | COL2A1 | 7q21.3–q22 | Cartilage, vitreous |
| III | COL3A1 | 12q13–q14 | Fetal skin, blood vessels, intestines |
| IV | COL4A1–6 | 13q34, 2q35–q37, Xq22 | Basement membranes |
| V | COL5A1–3 | 9q34.2–q34.3 | Ubiquitous |
| VI | COL6A1–3 | 21q22.3, 2q37 | Aortic intima, placenta |
| VII | COL7A1 | 3p21 | Amnion, anchoring fibrils |
| VIII | COL8A1–2 | 3q12–q13.1, 1p32.3–p34.3 | Endothelial cell cultures |
| IX | COL9A1–3 | 6q12–q14, 1p32 | Cartilage, type II collagen tissue |
| X | COL10A1 | 6q12–q22 | Cartilage |
| XI | COL11A1–2, COL2A1 | 1p21 | Cartilage, skin |
| XII | COL12A1 | 6 | Skin, cartilage, cornea, limbal |
| XIII | COL13A1 | 10q22 | Ubiquitous |
| XIV | COL14A1 | 8q23 | Ubiquitous, fetal hair follicles, basement membranes |
| XV | COL15A1 | 9q21–22 | Skin hemidesmosomes, kidney, liver, spleen |
| XVI | COL16A1 | 1p34 – 35 | Ubiquitous |
| XVII | COL17A1 | 10q24.3 | Skin hemidesmosomes (BP180) |
| XVIII | COL18A1 | 21q22.3 | Ubiquitous, basement membranes |
| XIX | COL19A1 | 6q12 – q14 | Ubiquitous, basement membranes |
| XX | COL20A1 | Corneal epithelium, embryonic skin, sternal cartilage, tendon | |
| XXI | COL21A1 | 6p11.2 – 12.3 | Blood vessel walls |
| XXII | COL22A1 | 8q24.2 | Tissue junctions such as basement membrane zone of anagen hair follicle |
| XXIII | Rat prostate carcinoma cells | ||
| XXIV | Fetal cornea and bone | ||
| XXV | Precursor to Alzheimer amyloid plaque component | ||
| XXVI | Testis, ovary | ||
| XXVII | Chondrocytes; developing tissues, including stomach, lung, gonad, skin, cochlea, teeth |
* A dash denotes a series of genes; e.g., COL14A1–2 indicates both the COL14A1 and the COL14A2 gene.
The regulation of collagen synthesis and degradation is complex. Dermal fibrosis is largely related to increases in type I collagen mediated by proα1 and proα2 collagen genes. Transforming growth factor–beta (TGF-β) results in increased type I procollagen synthesis. Angiotensin II type 1 receptor stimulation increases collagen production and inhibits collagen degradation, whereas type 2 receptor stimulation exerts the reverse effects. Mutations in collagens or disruption of their function due to autoimmunity or medications can lead to disease.
Czarny-Ratajczak M, et al: Collagens, the basic proteins of the human body. J Appl Genet 2000; 41: 317.
Elastosis Perforans Serpiginosa
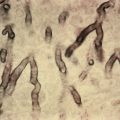
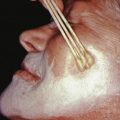
In 1953 Lutz described a chronic papular keratotic eruption in an arciform shape located on the sides of the nape of the neck. The papules range from 2–5 mm in diameter and are grouped in a serpiginous or horseshoe-shaped arrangement ( Fig. 25.1 ). The papules have a keratotic top that extrudes elastic fibers likely by binding to an elastin receptor on keratinocytes. Although the lesions typically occur on the neck, other sites may be involved, such as the upper arms, face, lower extremities, and rarely the trunk. Disseminated lesions may occur in Down syndrome. The disease runs a variable course, with spontaneous resolution often occurring from 6 months to 5 years after onset. Often, atrophic scarring remains. Progressive vasoocclusive disease with stroke has been reported.
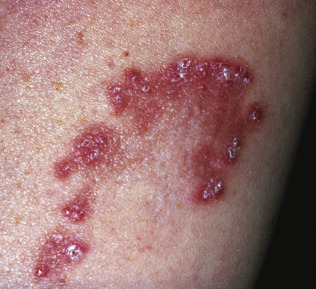
Elastosis perforans serpiginosa (EPS) is most common in young adults. Men outnumber women 4 : 1. Approximately one third of EPS cases occur in patients with associated diseases; the most common concomitant disorder is Down syndrome. Approximately 1% of patients with Down syndrome have EPS, and the lesions are likely to be more extensive and persistent than in other patients. EPS has also been reported in Ehlers-Danlos syndrome, osteogenesis imperfecta, Marfan syndrome, Rothmund-Thomson syndrome, acrogeria, systemic sclerosis, morphea, XYY syndrome, and renal disease. Reports of EPS associated with pseudoxanthoma elasticum have occurred with penicillamine administration. EPS can also be idiopathic. Evaluation for associated disease should be driven by associated signs and symptoms.
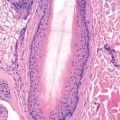
The distinctive histopathologic changes of EPS consist of elongated, tortuous channels in the epidermis into which eosinophilic elastic fibers perforate. The fibers are extruded from the dermis. There is degeneration and alteration of the elastic tissue in the adjacent papillary dermis with an accompanying inflammatory response. In penicillamine-associated disease, the fibers may have an irregular (bramble bush) contour when examined with electron microscopy.
Treatment of EPS is difficult, but individual lesions may resolve after liquid nitrogen cryotherapy. Some cases have responded to carbon dioxide (CO 2 ) (fractional in some reports), erbium:yttrium-aluminum-garnet (Er:YAG), or pulsed dye laser therapy. Topical retinoids and imiquimod have been reported to be of benefit.
Kelati A, et al: Treatment of elastosis perforans serpiginosa using a fractional carbon dioxide laser. JAMA Dermatol 2017; 153: 1063.
Kim SW, et al: A clinicopathologic study of thirty cases of acquired perforating dermatosis in Korea. Ann Dermatol 2014; 26: 162.
Lee SH, et al: Elastosis perforans serpiginosa. Ann Dermatol 2014; 26: 103.
Nasca MR, et al: Perforating pseudoxanthoma elasticum with secondary elastosis perforans serpiginosa-like changes. J Cutan Pathol 2016; 43: 1021.
Polańska A, et al: Elastosis perforans serpiginosa. Postepy Dermatol Alergol 2016; 33: 392.
Vearrier D, et al: What is standard of care in the evaluation of elastosis perforans serpiginosa? A survey of pediatric dermatologists. Pediatr Dermatol 2006; 23: 219.
Yang JH, et al: Treatment of elastosis perforans serpiginosa with the pinhole method using a carbon dioxide laser. Dermatol Surg 2011; 37: 524.
Reactive Perforating Collagenosis
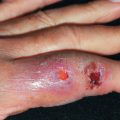
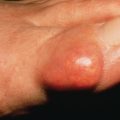
In 1967 Mehregan reported a rare, familial, nonpruritic skin disorder characterized by papules that grow to a diameter of 4–6 mm and develop a central area of umbilication in which keratinous material is lodged. The discrete papules may be numerous ( Fig. 25.2 ) and involve sites of frequent trauma, such as the backs of the hands, the forearms, elbows, and knees. The lesion reaches a maximum size of about 6 mm in 4 weeks and then regresses spontaneously in 6–8 weeks. The lesions are broader than those of EPS, and a broad crust containing collagen fibers is extruded centrally. Koebnerization is often observed. Young children are most frequently affected. Most reports support an autosomal recessive mode of inheritance, although apparent autosomal dominant inheritance was reported in one family but the genetics have not been elucidated. Acquired reactive perforating collagenosis is discussed further in Chapter 33 .
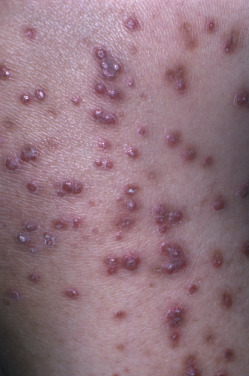
No specific treatment is typically indicated for reactive perforating collagenosis because the lesions involute spontaneously. Topical retinoids may be helpful in patients who require treatment.
Ramesh V, et al: Familial reactive perforating collagenosis. J Eur Acad Dermatol Venereol 2007; 21: 766.
Tiwary AK, et al: A rare case of familial reactive perforating collagenosis. Indian J Dermatol 2017; 18: 230.
Pseudoxanthoma Elasticum
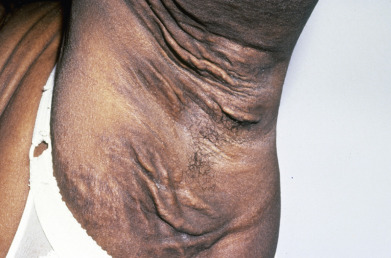
Pseudoxanthoma elasticum (PXE) is an inherited disorder involving the connective tissue of the skin, eye, and cardiovascular system. Many cases are sporadic. In familial cases, both a recessive and a dominant inheritance pattern have been reported, with the recessive form apparently more common. The skin changes generally present as small, circumscribed, yellow to cream-colored papules on the sides of the neck and flexures, giving the skin a “plucked chicken skin” appearance ( Fig. 25.3 ). Lax, redundant folds of skin may be present ( Fig. 25.4 ). Nuchal comedones and milia en plaque may also be seen. Characteristic exaggerated nasolabial folds and mental creases are common. Mental creases (horizontal creases across the chin) appearing in patients under age 30 are highly suggestive of PXE. In addition, the inguinal, periumbilical, and periauricular skin, as well as the mucosa of the soft palate, inner lip, tonsils, stomach, rectum, and vagina, may be involved.
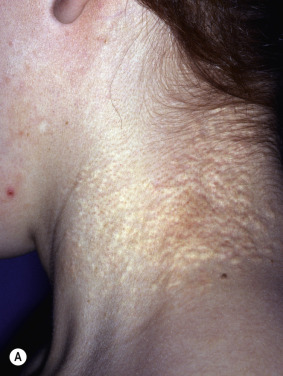
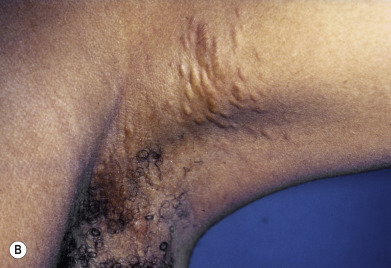
The characteristic retinal change is the angioid streak, which is the result of breaks in Bruch’s elastic membrane. PXE can be demonstrated in more than half of patients with angioid streaks, and 85% of PXE patients will have retinal findings. The angioid streaks appear earlier than the skin changes, so most cases are discovered by ophthalmologists. Angioid streaks may be the only sign of the disease for years. Biopsies of the midportions of old scars may be diagnostic of PXE. Angioid streaks may also be seen in Ehlers-Danlos syndrome, Paget disease of bone, diabetes, hemochromatosis, hemolytic anemia, hypercalcinosis, solar elastosis, neurofibromatosis, Sturge-Weber syndrome, tuberous sclerosis, myopia, sickle cell anemia, trauma, lead poisoning, hyperphosphatemia, pituitary disorders, and intracranial disorders. PXE, Paget disease of the bone, and sickle cell disease account for the vast majority of patients with angioid streaks.
On funduscopic examination, a reddish brown band is evident around the optic disk, from which glistening streaks extend. In addition, there may be hemorrhages and exudates. Progressive loss of vision often starts after minor trauma to the eye. Drusen-like spots are often present and show increased autofluorescence, unlike age-related drusen.
Vascular involvement frequently leads to hemorrhage. These vascular events are caused by the degeneration of the elastic fibers in the vascular media. Gastric hemorrhage occurs in 10% of patients, and on gastroscopy, diffuse bleeding is common. Epistaxis occurs frequently, but hematuria is rare. PXE affects the elastic tissue of the cardiac valves, myocardium, and pericardium. In one study, mitral valve prolapse was found in 71% of 14 patients examined. Hypertension occurs in many patients older than age 30. Any patient with hypertension at a young age should be examined for stigmata of PXE. Leg cramps and intermittent claudication occur prematurely, and peripheral pulses are diminished or absent. Calcification of peripheral arteries is seen in many patients over age 30 and may be detected by radiography. Accelerated coronary artery disease (CAD) can occur, especially in association with hypertension. Extensive cutaneous calcification and renal and testicular stones may occur.
Mutations in the adenosine triphosphate (ATP)–binding cassette transporter protein subfamily C member 6 gene (ABCC6) have been implicated in the pathogenesis of PXE in a majority of patients, who also have a higher incidence of CAD. Defective release of ATP leads to less inorganic pyrophosphate (PPi), which is important in inhibiting mineralization; thus the mineralization goes unchecked. Although the most prominent manifestations of the disease are in the skin, eye, gut, and heart, mineralization of elastic fibers can be found in many organs.
Histologically, elastic fibers are fragmented and mineralized with calcium. The fibers stain gray-blue with hematoxylin and eosin (H&E) and are twisted, curled, and broken, suggesting “raveled wool.” Blind biopsies of scars or axillary skin in patients with a family history of PXE or with angioid streaks may show early changes of PXE. Calcium stains are helpful in identifying early disease.
The differential diagnosis includes PXE-like papillary dermal elastolysis, perforating calcific elastosis, connective tissue nevus with elastorrhexis, and cutis laxa. Patients with PXE-like papillary dermal elastolysis may have cobblestoned, yellow papules on the neck, similar to PXE, but lack any retinal or vascular alterations and the typical fragmentation of elastic fibers with calcium deposition on histology. Patients described with connective tissue nevus with elastorrhexis have tiny white papules coalescing into a plaque on the upper chest and lower neck that are not associated with any internal findings. Penicillamine may induce similar clinicohistologic features in patients with Wilson disease or homocystinuria.
No definitive therapy is available to treat the skin disease. Some data suggest that PXE patients benefit from limiting dietary calcium and phosphorus to the minimal daily requirement. Intravitreal bevacizumab has been used to treat choroidal neovascularization. Atorvastatin treatment appears promising in a mouse model. Therapeutic trials of magnesium and a bisphosphonate are ongoing.
Chu DH, et al: A new variant of connective tissue nevus with elastorrhexis and predilection for the upper chest. Pediatr Dermatol 2015; 32: 518.
Decani S, et al: Pseudoxanthoma elasticum of the palate. Oral Surg Oral Med Oral Pathol Oral Radiol 2016; 121: e6.
Finger RP, et al: Intravitreal bevacizumab for choroidal neovascularisation associated with pseudoxanthoma elasticum. Br J Ophthalmol 2008; 92: 483.
Guo H, et al: Atorvastatin counteracts aberrant soft tissue mineralization in a mouse model of pseudoxanthoma elasticum (Abcc6−/−). J Mol Med (Berl) 2013; 91: 1177.
Hendig D, et al: New insights into the pathogenesis of pseudoxanthoma elasticum and related soft tissue calcification disorders by identifying genetic interactions and modifiers. Front Genet 2013; 4: 114.
Plomp AS, et al: Proposal for updating the pseudoxanthoma elasticum classification system and a review of the clinical findings. Am J Med Genet 2010; 152A: 1049.
Uitto J, et al: Insights into pathomechanisms and treatment development in heritable ectopic mineralization disorders. J Invest Dermatol 2017; 137: 790.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


