Amyloidosis
Amyloid is a material deposited in the skin and other organs that is eosinophilic, homogeneous, and hyaline in appearance. It represents beta-pleated sheet forms of various host-synthesized molecules processed into this configuration by host cells.
Amyloidosis can be classified as systemic, localized, and heredofamilial types. The systemic types can deposit amyloid in multiple organs due to an overproduction of a host protein that cannot be adequately excreted or metabolized by the host. The excess protein is metabolized into amyloid precursors that interact with tissue proteoglycans/glycosaminoglycans, forming soluble amyloid oligomers. These oligomers complex with serum amyloid P (SAP), forming amyloid deposits in the affected organ. Over 30 proteins have been associated with amyloidosis, including immunoglobulin light chains (myeloma associated amyloid), tranthyretin (senile amyloid), beta-2 microglobulin (diabetic amyloid), and alpha natriuretic protein. In all forms of amyloid, the pattern of deposition is characteristic, although there can be overlap between various forms. The diagnosis of a specific type of amyloid should only be made if the clinical features are characteristic and if the deposited protein is identified histochemically. Primary localized amyloidosis (also called primary cutaneous amyloidosis when the skin is affected) is very common. Rare familial syndromes may be complicated by secondary systemic amyloidosis or may have genetic defects that lead to amyloid deposition (heredofamilial amyloidosis). Classification of cutaneous amyloidoses is as follows:
- I.
Systemic amyloidosis
- A.
Primary (myeloma-associated) systemic amyloidosis
- B.
Secondary systemic amyloidosis
- C.
Dialysis-related amyloidosis
- D.
Senile systemic amyloidosis
- A.
- II.
Cutaneous amyloidosis
- A.
Macular amyloidosis
- B.
Lichen amyloidosis
- C.
Nodular amyloidosis
- D.
Secondary (tumor-associated) cutaneous amyloidosis
- E.
Familial primary cutaneous amyloidosis
- F.
Pharmaceutical amyloidosis
- A.
- III.
Heredofamilial amyloidosis
All forms of amyloid have relatively identical histologic and electron microscopic findings. The amyloid in all forms is made up of three distinct components: protein-derived amyloid fibers, amyloid P component (about 15% of amyloid), and ground substance. The protein-derived amyloid fibers are those that differ among the various forms of amyloid.
Amyloid is weakly periodic acid–Schiff (PAS) positive and diastase resistant, Congo red positive, purple with crystal violet, and positive with thioflavin T. Amyloid stained with Congo red exhibits apple-green birefringence under polarized light. Secondary systemic amyloid (AA amyloid) loses its birefringence after treatment with potassium permanganate, whereas primary and localized cutaneous forms do not.
Amyloid stains an intense, bright orange with cotton dyes such as Dylon, Pagoda red, RIT Scarlet No. 5, or RIT Cardinal Red No. 9. Ultrastructurally, amyloid has a characteristic fibrillar structure that consists of straight, nonbranching, nonanastomosing, often irregularly arranged filaments 60–100 nm in diameter. In most cases, specific antibodies against the protein component should be used to confirm the type of amyloidosis. Because amyloid substance P is present in all forms of amyloid, immunoperoxidase staining against this component will stain all forms of amyloid. In addition, because SAP is avidly bound to amyloid, radiolabeled, highly purified SAP can be used to localize amyloidosis, determine the extent of organ infiltration, study progression of disease, and determine whether therapy reduces the amount of amyloid in various organs. Radiolabeled SAP scintigraphy and identification of specific amyloid proteins immunohistochemically can be done at some specialized centers, although the amount of deposition is not directly correlated with organ dysfunction.
Systemic Amyloidoses
Primary Systemic Amyloidosis (AL Amyloidosis)
Primary systemic amyloidosis typically involves the kidneys, liver, heart, gastrointestinal (GI) tract, peripheral nerve tissue, and skin. Myeloma-associated amyloidosis is included in this category. The amyloid fibril proteins in primary systemic amyloidosis are composed of the protein AL amyloid, a portion of the immunoglobulin (Ig) light chain. It is usually of the lambda (λ) subtype, and certain germline Ig light-chain V chains (6aVλVI and 3rVλIII) are responsible for AL amyloidosis in 40% of patients.
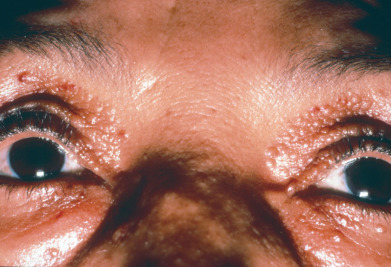
Cutaneous manifestations occur in approximately 40% of patients with primary systemic amyloidosis. The cutaneous eruption usually begins as shiny, smooth, firm, flat-topped, or spherical papules of waxy color that have the appearance of translucent vesicles. These lesions coalesce to form nodules and plaques of various sizes and, in some cases, bandlike lesions. The regions around the eyes, nose, mouth, and mucocutaneous junctions are frequently involved ( Fig. 26.1 ). Vulvar lesions may resemble giant condylomata. Lesions may also be uniform small papules resembling milia or even microcystic lymphatic malformations. Follicular plugging may occur, resulting in milia.
Purpuric lesions and ecchymoses occur in about 15% of patients and are the most common cutaneous manifestation of primary systemic amyloidosis. There are several mechanisms by which AL amyloid leads to purpura. Amyloid may infiltrate blood vessels, making them fragile. AL amyloid may also bind factor X, inhibiting its function and thus leading to pupura. Last, amyloid infiltration of the liver may lead to reduced production of fibrinogen and factor X, adversely affecting clotting. Purpura chiefly involves the eyelids, limbs, and oral cavity. It typically occurs after trauma (pinch purpura) and can be reproduced by the physician by rubbing a pen or dull instrument over the skin, analogous to trying to demonstrate dermatographism. Purpuric lesions also classically appear after actions or procedures that result in increased pressure in the vessels of the face, such as after vomiting, coughing, proctoscopic examination, or pulmonary function testing.
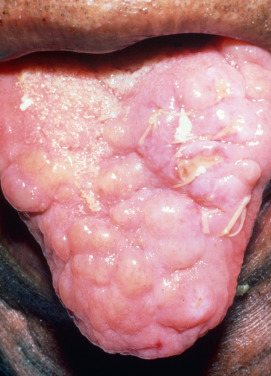
Glossitis, with macroglossia, occurs in at least 20% of patients, may be an early symptom, and can lead to dysphagia. The tongue becomes greatly enlarged, and furrows develop. The lateral aspects show indentations from the teeth. Papules or nodules, sometimes with hemorrhage, occur on the tongue ( Fig. 26.2 ).
Bullous amyloidosis is a rare but important clinical manifestation of amyloidosis. Skin fragility and tense, hemorrhagic or clear, noninflammatory bullae appear at areas of trauma, usually the hands, forearms, and feet. Lesions heal with scarring and milia. The esophagus and oropharyngeal mucosae may also be involved. Histologically, the lesions are subepidermal and pauci-inflammatory. Epidermolysis bullosa acquisita and porphyria cutanea tarda are in the differential diagnoses because they both cause blisters that heal with milia. Amyloid staining may yield negative results, and direct immunofluorescence (DIF) may be falsely positive because of AL protein deposition at the dermoepidermal junction (DEJ). The diagnosis is confirmed by evaluation of the patient’s serum and urine for Ig fragments and by amyloid stains or electron microscopy of the skin biopsies.
A diffuse or patchy alopecia, cutis verticis gyrata, and a scleroderma-like, scleromyxedema-like, or a cutis laxa–like appearance have also rarely been described. Cutis laxa–like findings may be generalized or localized to the acral parts. Lesions in the flexors and lateral neck may resemble pseudoxanthoma elasticum (PXE). At times, lesions with cutis laxa–like or PXE-like appearance may show amyloid bound to elastic fibers. The nail matrix may be infiltrated, resulting in atrophy of the nail plate, presenting as longitudinal striae, partial anonychia, splitting, and crumbling of the nail plate. Cordlike thickening along blood vessels can also occur. Bilateral stenosis of the external auditory canals has been reported. Patients with systemic amyloidosis are at increased risk for skin cancer.
Patients may present with or develop a plethora of systemic findings. Most characteristically, they develop carpal tunnel syndrome, other peripheral neuropathies, a rheumatoid arthritis (RA)–like arthropathy of the small joints, orthostatic hypotension, GI bleeding, nephrotic syndrome, and cardiac disease.
About 90% of patients will have the Ig fragment detectable in the serum or urine; in the other 10%, the serum free light-chain assay will detect a clear excess of one of the light chains (κ or λ), confirming the diagnosis. Also, reduction of the urine free light chains by more than 50% correlates with substantial benefit from treatment. Cardiac troponins are elevated and are powerful prognostic determinants in AL amyloidosis. Elevated troponins are associated with a 6-month survival. AL patients may appear to have prominent deltoid muscles as a result of deposition of amyloid in the muscles (shoulder pad sign). Cardiac arrhythmias and right-sided congestive heart failure are common causes of death.
The prognosis for patients with primary systemic amyloidosis is poor, and therapy is targeted toward decreasing the production of the initial protein that makes the amyloid. Those presenting with neurologic findings survive longer than patients presenting with cardiac disease. Approximately 15% of patients with AL amyloidosis will have myeloma, and 15% of patients with myeloma will have AL amyloidosis.
Secondary Systemic Amyloidosis (AA Amyloidosis)
Secondary systemic amyloidosis is caused by a chronic infectious or inflammatory process. In these conditions, the precursor protein, serum amyloid A (SAA), an acute-phase reactant, is chronically elevated and cannot be adequately cleared from the body. It is processed to AA amyloid in affected tissues. With more effective treatment for chronic infections (especially tuberculosis, schistosomiasis, osteomyelitis, bronchiectasis, pyelonephritis, and decubitus ulcer), infection-related AA amyloid is much less common. Most cases are now related to chronic inflammatory conditions, especially RA, juvenile idiopathic arthritis, ankylosing spondylitis, adult Still disease, inflammatory bowel disease, and Behçet disease. Amyloidosis has also been reported in alkaptonuria. The newer and more aggressive management strategies for these inflammatory conditions have led to reduced numbers or delayed onset of AA amyloidosis in these patients. Maintaining SAA below 4 mg/L is associated with a good outcome in AA amyloidosis. The organs involved most commonly by AA amyloidosis are the kidneys, adrenals, liver, and spleen. The skin is not involved, but biopsy of skin in patients with AA amyloidosis will detect amyloid deposits in the dermis perivascularly. Certain skin conditions, such as hidradenitis suppurativa, stasis ulcers, psoriatic arthritis, and dystrophic epidermolysis bullosa, may be complicated by AA amyloidosis. Many inherited conditions associated with elevated SAA may be complicated by AA amyloidosis as well. These include familial Mediterranean fever, cryopyrin-associated periodic syndromes, and tumor necrosis factor (TNF) receptor–associated periodic syndrome (TRAPS).
Dialysis-Associated Amyloidosis (β 2 -Microglobulin Amyloidosis)
β 2 -Microglobulin is excreted primarily by the kidneys. In patients with severe renal failure on dialysis or predialysis, the excess β 2 -microglobulin may be processed to amyloid in certain tissues. Almost 100% of patients receiving dialysis for 15 years or more will develop this form of amyloidosis. It primarily affects the synovium, causing musculoskeletal symptoms, carpal tunnel syndrome, and, less often, triggers finger, bone cysts, and spondyloarthropathy. Rarely subcutaneous tumors can arise often of the buttocks overlying the sacrum. Pedunculated sacral masses, lichenoid papules, and localized hyperpigmentation can also be seen. The diagnosis is confirmed by biopsy, which demonstrates that the amyloid material is β 2 -microglobulin on immunohistochemical stains. The treatment is high-flux dialysis or kidney transplantation.
Senile Systemic Amyloidosis
Senile systemic amyloidosis is increasingly recognized as an important cause of cardiac disease in the elderly population (>70 years). Carpal tunnel syndrome can also occur. Senile systemic amyloidosis is caused by deposition of normal transthyretin, a transporter protein, in tissue. Skin lesions have not been reported, but vascular deposition has led to tongue necrosis. The diagnosis can be confirmed in about three quarters of patients with a deep abdominal fat biopsy.
Cutaneous Amyloidosis
Primary Localized Cutaneous Amyloidosis
The primary localized cutaneous amyloidoses have been divided into four forms: macular, lichen, nodular, and familial. Macular and lichen forms of amyloidosis are also called “keratinocyte-derived” amyloidosis, frictional amyloidosis, and frictional melanosis. Some cases of these two forms of cutaneous amyloidosis are familial, but the relationship between these and cases of familial primary localized cutaneous amyloidosis is unclear. Patients with macular and lichen amyloidosis often have coexistent atopic dermatitis and the scratching is likely the cause of the amyloid. Nodular and familial cases of cutaneous amyloidosis are rare and have a unique pathogenesis.
Nonfamilial macular and lichen amyloidosis have the same pathogenic basis (rubbing and friction), and overlap cases (biphasic cutaneous amyloidosis) can be seen. Individuals of Asian, Hispanic, or Middle Eastern ancestry seem to be predisposed and use of abrasive devices during bathing is a precipitant. In cases of acquired macular and lichen amyloidosis, the deposited amyloid material contains keratin (primarily keratin 5) as its protein component, strongly suggesting that traumatic damage to basal keratinocytes results in the deposits. A rare form localized to the conchae has been described. Many pruritic skin conditions such as primary biliary cirrhosis and chronic renal failure have been associated.
The histologic picture of acquired macular and lichen amyloidosis is similar; the only difference is the size of the amyloid deposits and the extent of the overlying epidermal changes. The overlying epidermis is frequently hyperkeratotic and focally acanthotic, as a result of the chronic rubbing. Focal necrotic keratinocytes may be observed in the basal cell layer. Microscopic and rarely macroscopic bullae (analogous to those in lichen planus) may be seen. Dermal papillae are expanded by amorphous deposits of amyloid that abut immediately below the epidermis. Melanin deposits are classically present in the amyloid. In cases of postinflammatory hyperpigmentation with incontinence of pigment, the architecture of the areas of dermal melanosis should be examined carefully to exclude amyloidosis. Systemic amyloidosis is excluded by the absence of amyloid deposits around blood vessels. Special stains may be used to confirm the diagnosis, but this is rarely required if the classic histology is found. In difficult cases, immunoperoxidase for keratin will stain the amyloid deposits and confirm the diagnosis of primary cutaneous amyloidosis. DIF may demonstrate immunoglobulin (usually IgM) in a globular pattern in the keratin-derived cutaneous amyloidoses, but this is caused by passive absorption rather than specific deposition. This phenomenon is seen in all disorders with prominent apoptosis of keratinocytes.
Macular Amyloidosis
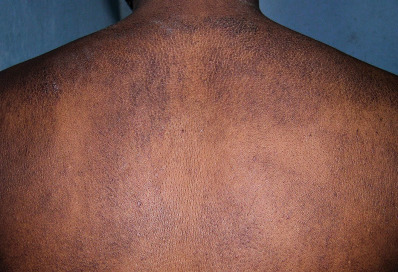
Typically, patients with macular amyloidosis exhibit moderately pruritic, brown, rippled macules characteristically located in the interscapular region of the back ( Fig. 26.3 ). Women outnumber men by 5 : 1 and the disease is chronic. Pigmentation is generally not uniform, giving the lesions a “salt and pepper” or rippled appearance. Many cases of macular amyloid between the scapulae probably result from rubbing dysesthetic areas of notalgia paresthetica. Occasionally, the thighs, shins, arms, breasts, and buttocks may be involved, and these more diffuse cases are usually associated with diffuse pruritus.
Lichen Amyloidosis
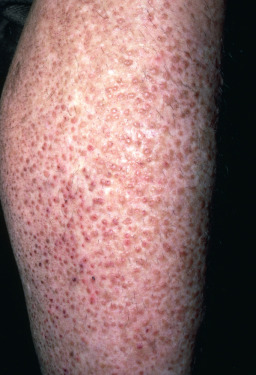
Lichen amyloidosis is characterized by the appearance of paroxysmally itchy lichenoid papules, virtually always appearing bilaterally on the shins ( Fig. 26.4 ). Some patients may deny itching but there is still chronic rubbing. Men outnumber women 2 : 1. Lichen amyloidosis may be found in approximately one third of patients with multiple endocrine neoplasia type IIA (see later discussion). The primary lesions are small, brown, discrete, slightly scaly papules that group to form large, infiltrated plaques. Less frequently, these may occur on the thighs, forearms, face, and even the upper back.
Treatment
Treatment of lichen and macular cutaneous amyloidosis is frequently unsatisfactory. Reducing friction is critical. Identifying the cause of the rubbing, and whether it is habit, pruritus, or neuropathy (as in notalgia paresthetica), directs treatment. Occlusion of therapy is helpful because it both enhances topical treatments and provides a physical block to prevent trauma to the skin. High-potency topical corticosteroids can be beneficial, as can intralesional corticosteroid therapy when small areas are involved. Topical tacrolimus 0.1% ointment, psoralen plus ultraviolet A light (PUVA) and with retinoids (Re-PUVA), ultraviolet B (UVB) light, tar, and calcipotriol benefit individual patients. Amitriptyline (for itching), oral retinoids, thalidomide, and systemic immunosuppressants, including corticosteroids, may be used in refractory cases. The pigmentation of macular amyloidosis reportedly has been improved by laser therapy, especially the 532-nm Q-switched neodymium-doped yttrium-aluminum-garnet (Nd:YAG) laser.
Nodular Amyloidosis
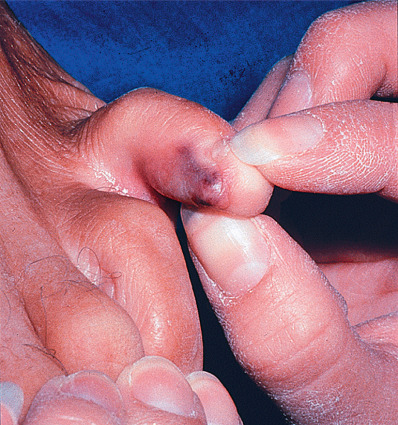
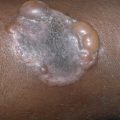
Nodular amyloidosis is a rare form of primary localized cutaneous amyloidosis in which single or, less often, multiple nodules or tumefactions preferentially involve the acral areas ( Fig. 26.5 ). However, trunk, genital, or facial lesions may be seen as well. The lesions are asymptomatic, vary in size from several millimeters to several centimeters, and may grow slowly after their initial appearance. The overlying epidermis may appear atrophic, and lesions may resemble large bullae. Numerous conditions have been associated with nodular primary localized cutaneous amyloidosis (NPLCA), especially Sjögren syndrome, but also systemic sclerosis (including CREST) and RA. In Sjögren syndrome the nodular amyloidosis typically appears around age 60, more frequently in females, and may precede the diagnosis of Sjögren syndrome by many years. The dermis and subcutis may be diffusely infiltrated with amyloid. The lesions may contain numerous plasma cells and may be early MALT lymphomas. The amyloid in these patients is Ig-derived AL, as is seen in primary systemic amyloidosis, and is unrelated to keratinocyte-related amyloid or to AA amyloid. Progression to systemic amyloidosis may occur in about 7% of cases, so they should be regularly evaluated for progression. Treatment is physical removal or destruction of the lesion with shave removal and destruction of the base.
Secondary Cutaneous Amyloidosis
After PUVA therapy and in benign and malignant cutaneous neoplasms, deposits of amyloid may be found. Most frequently, the associated neoplasms are nonmelanoma skin cancers or seborrheic keratoses. Discoid lupus, dermatomyositis (DM), and graft-versus-host disease (GVHD), as interface dermatoses with apoptosis of keratinocytes, can occasionally demonstrate amyloid in the upper dermis. In all cases, this is keratin-derived amyloid.
Hereditary Cutaneous Amyloidosis Syndromes
Familial primary localized cutaneous amyloidosis (FPLCA) is an autosomal dominant syndrome associated with chronic itching (although some deny it) and cutaneous lesions resembling macular and lichen amyloidosis. It is seen most often in Japan, Brazil, China, and Taiwan. The age of onset is 5–18 years. In some families, sun exposure may be an exacerbating factor. Lesions are often widespread on the limbs, chest, and upper and lower back. The buttocks, conchae, and dorsal feet and hands may also be involved. Mutations in the OSMRβ or interleukin-31 receptor A (IL-31RA) genes that code for proteins that form the two subunits of the transmembrane receptor for IL-31. IL-31 induces the secretion of monocyte chemotactic protein 1 (MCP-1), and levels of MCP-1 expression are very low in FPLCA. MCP-1 recruits monocytes to clear the cellular debris resulting from keratinocyte damage. In the absence of this signal, cellular debris accumulates, and the keratin is processed to amyloid. There is hypersensitivity of small nerve fibers leading to itching. Rare cases of macular amyloidosis in a blaschkoid distribution suggest that mosaicism for FPLCA can be seen, giving this unusual cutaneous distribution. Some FPLCA cases demonstrate extensive poikilodermatous lesions, and less frequently a patient may have multiple morphologies of lichen amyloid, poikiloderma, and dyschromica and even small bullous lesions.
Amyloidosis cutis dyschromica is a distinct type of FPLCA with onset in childhood, no pruritus, a dotted reticular hyperpigmentation with hypopigmented spots without papulation covering almost all the body, and small foci of amyloid just below the epidermis. The nature of the amyloid is unclear. Most affected families are from Japan, Taiwan, and India. UVB hypersensitivity is often reported by these patients.
Multiple endocrine neoplasia type IIA (MEN-2A) syndrome and familial medullary thyroid carcinoma (FMTC) are both caused by mutations in the RET proto-oncogene. Cutaneous amyloidosis, most often keratin-derived macular amyloidosis, may be seen in these patients. The macular amyloid may be restricted to the upper back and also unilateral (associated with notalgia paresthetica), or it may be bilateral and more extensive. Age of onset is usually before 20. Thirty of 31 patients with MEN-2A had cutaneous amyloidosis before the diagnosis of MEN-2A was made. In a patient with macular amyloidosis of early onset (before age 20), a careful family history should be taken for endocrine neoplasias, the skin and mucosa examined for neuromas, the blood pressure taken (checking for pheochromocytoma), and the thyroid palpated. A serum calcitonin level should be ordered and, if elevated, a thyroid ultrasound performed.
Pharmaceutical Amyloidosis
When injected into the skin, insulin can create deposits of amyloid composed of the A and B subunits of insulin. This is termed AIns. Lesions present as deep subcutaneous nodules, usually on the lower abdomen. If patients inject into these sites, their glucose control may be impaired as their insulin is less effective. Injecting into new areas or surgically removing the nodules improves glucose control. Enfuvirtide, a human immunodeficiency virus (HIV) fusion–inhibiting peptide administered subcutaneously, can produce similar lesions.
Familial Syndromes Associated With Amyloidosis (Heredofamilial Amyloidosis)
Most forms of familial amyloidosis are caused by abnormal host proteins that cannot be adequately processed, resulting in their deposition in various tissues in the form of amyloid. Only 50% of patients with hereditary amyloidosis will have a positive family history. Liver, kidney, heart, eye, and nervous system may be involved. Several types of hereditary amyloidosis have been identified; some forms are caused by genetic defects in transthyretin. These are autosomal dominant syndromes, and most affected patients are heterozygotes. Others are caused by a genetic defect in apolipoprotein A-I or A-II, by a defect in gelsolin, fibrinogen A-α, cystatin C, or lysozyme. These syndromes must often be diagnosed by genetic testing or immunohistochemical identification of the deposited pathogenic protein.
Alvarez-Ruiz SB, et al: Unusual clinical presentation of amyloidosis. Int J Dermatol 2007; 46: 503.
An Q, et al: Thalidomide improves clinical symptoms of primary cutaneous amyloidosis. Dermatol Ther 2013; 26: 263.
Andoh T, et al: Increase in sensory sensitivity around, but not in the central part of, the hyperkeratotic papule in lichen amyloidosis. Br J Dermatol 2017; ePub ahead of print.
Arnold SJ, Bowling JC: “Shiny white streaks” in lichen amyloidosis. Australas J Dermatol 2012; 53: 272.
Barja J, et al: Systemic amyloidosis with an exceptional cutaneous presentation. Dermatol Online J 2013; 19: 11.
Chandran NS, et al: Case of primary localized cutaneous amyloidosis with protean clinical manifestations. J Dermatol 2011; 38: 1066.
Chu H, et al: Successful treatment of lichen amyloidosis accompanied by atopic dermatitis by fractional CO 2 laser. J Cosmet Laser Ther 2017; 19: 345.
Clos AL, et al: Role of oligomers in the amyloidogenesis of primary cutaneous amyloidosis. J Am Acad Dermatol 2011; 65: 1023.
De Sousa SM, McCormack AI: Cutaneous lichen amyloidosis in multiple endocrine neoplasia. Intern Med J 2016; 46: 116.
D’Souza A, et al: Pharmaceutical amyloidosis associated with subcutaneous insulin and enfuvirtide administration. Amyloid 2014; 21: 71.
Garg T, et al: Amyloidosis cutis dyschromica. J Cutan Pathol 2011; 38: 823.
Grimmer J, et al: Successful treatment of lichen amyloidosis with combined bath PUVA photochemotherapy and oral acitretin. Clin Exp Dermatol 2007; 32: 39.
Haemel A, et al: Keratinocyte-derived amyloidosis as a manifestation of chronic graft-versus-host disease. J Cutan Pathol 2013; 40: 291.
Hanami Y, Yamamoto T: Secondary amyloid deposition in a melanocytic nevus. Int J Dermatol 2013; 52: 1031.
Hemminki K, et al: Cancer risk in amyloidosis patients in Sweden with novel findings on non-Hodgkin lymphoma and skin cancer. Ann Oncol 2014; 25: 511.
Kalkan G, et al: An alternative treatment model: the combination therapy of narrow band ultraviolet B phototherapy and tacrolimus ointment 0.1% in biphasic amyloidosis. J Pak Med Assoc 2014; 64: 579.
Kandhari R, et al: Asymptomatic conchal papules. Indian J Dermatol Venereol Leprol 2013; 79: 445.
Koh M, et al: A rare case of primary cutaneous nodular amyloidosis of the face. J Eur Acad Dermatol Venereol 2008; 22: 1011.
Konopinski JC, et al: A case of nodular cutaneous amyloidosis and review of the literature. Dermatol Online J 2013; 19: 10.
Kumar S, et al: Skin involvement in primary systemic amyloidosis. Mediterr J Hematol Infect Dis 2013; 5: e2013005.
Kurian SS, et al: Amyloidosis cutis dyschromica. Indian Dermatol Online J 2013; 4: 344.
Kuseyri, O et al: Amyloidosis cutis dyschromica, a rare cause of hyperpigmentation. Pediatrics 2017; 139: e20160170.
LaChance A, et al: Nodular localized primary cutaneous amyloidosis. Clin Exp Dermatol 2014; 39: 344.
Lin JR, et al: Tongue necrosis and systemic vascular amyloidosis. Human Pathol 2011; 42: 734.
Love WE, et al: The spectrum of primary cutaneous nodular amyloidosis. J Am Acad Dermatol 2008; 58: S33.
Meijer JM, et al: Sjögren’s syndrome and localized nodular cutaneous amyloidosis. Arthritis Rheum 2008; 58: 1992.
Merchand A, et al: Cutaneous amyloid elastosis revealing multiple myeloma with systemic amyloidosis. Acta Derm Venereol 2013; 93: 204.
Millucci L, et al: Diagnosis of secondary amyloidosis in alkaptonuria. Diagn Pathol 2014; 9: 185.
Mohty D, et al: Cardiac amyloidosis. Arch Cardiovasc Dis 2013; 106: 528.
Nagaste T, et al: Insulin-derived amyloidosis and poor glycemic control Am J Med 2014; 127: 450.
Ostovari N, et al: 532-nm and 1064-nm Q-switched Nd:YAG laser therapy for reduction of pigmentation in macular amyloidosis patches. J Eur Acad Dermatol Venereol 2008; 22: 442.
Pardo Arranz L, et al: Familial poikylodermic cutaneous amyloidosis. Eur J Dermatol 2008; 18: 289.
Park MY, Kim YC: Macular amyloidosis with an incontinentia pigmenti–like distribution. Eur J Dermatol 2008; 18: 477.
Renker T, et al: Systemic light-chain amyloidosis revealed by progressive nail involvement, diffuse alopecia and sicca syndrome. Dermatology 2014; 228: 97.
Ritchie SA, et al: Primary localized cutaneous nodular amyloidosis of the feet. Cutis 2014; 93: 89.
Rothberg AE, et al: Familial medullary thyroid carcinoma associated with cutaneous lichen amyloidosis. Thyroid 2009; 19: 651.
Sakuma TH, et al: Familial primary localized cutaneous amyloidosis in Brazil. Arch Dermatol 2009; 145: 695.
Salim T, et al: Lichen amyloidosis. Indian J Dermatol Venereol Leprol 2005; 71: 166.
Shiao YM, et al: MCP-1 as an effector of IL-31 signaling in familial primary cutaneous amyloidosis. J Invest Dermatol 2013; 133: 1375.
Susantitaphong P, Dember LM, Jaber BL: Dialysis-associated amyloidosis. In: Picken M, Herrera G, Dogan A (Eds.), Current Clinical Pathology: Amyloid and Related Disorders (pp. 81-94). New York: 2015, Humana Press.
Taniquchi Y, et al: Cutaneous amyloidosis associated with amyopathic dermatomyositis. J Rheumatol 2009; 36: 1088.
Tey HL, et al: Pathophysiology of pruritus in primary localized cutaneous amyloidosis. Br J Dermatol 2016; 174: 1345.
Tong PL, et al: Primary localized cutaneous nodular amyloidosis successfully treated with cyclophosphamide. Australas J Dermatol 2013; 54: e12.
Walsh N, et al: Sjogren’s syndrome and localized nodular cutaneous amyloidosis. Lupus Sci Med 2017; 4.
Wang WH, et al: A new c.1845A→T of oncostatin M receptor-β mutation and slightly enhanced oncostatin M receptor-β expression in a Chinese family with primary localized cutaneous amyloidosis. Eur J Dermatol 2012; 22: 29.
Wat H, et al: Primary systemic (amyloid light-chain) amyloidosis masquerading as pseudoxanthoma elasticum. JAMA Dermatol 2014; 150: 1091.
Wechalekar AD, et al: Systemic amyloidosis. Lancet 2016; 387: 2641.
Weidner T, et al: Primary localized cutaneous amyloidosis. Am J Clin Dermatol 2017; 18: 629.
Yang W, et al: Amyloidosis cutis dyschromica in two female siblings. BMC Dermatol 2011; 11: 4.
Yasuyuki F, et al: Nail dystrophy and blisters as sole manifestations in myeloma-associated amyloidosis. J Am Acad Dermatol 2006; 54: 712.
Yew YW, Tey HL: Itch in familial lichen amyloidosis. Dermatol Ther 2014; 27: 12.
Zhao JY, et al: A case of systemic amyloidosis beginning with purpura. Chin Med J 2012; 125: 555.
Porphyrias
Porphyrinogens are the building blocks of all the hemoproteins, including hemoglobin and the cytochrome enzymes, and are produced primarily in the liver and bone marrow. Each form of porphyria has now been associated with a deficiency in an enzyme in the metabolic pathway of heme synthesis. These enzyme deficiencies lead to accumulation of the precursor molecules before the mutation. The precursors are porphyrins, and the diseases are called porphyrias. Many of the porphyrins lead to severe photosensitivity that accounts for most of the cutaneous findings.
Understanding the biosynthetic pathway of heme has clarified the biochemical basis of the porphyrias. Delta-aminolevulinic acid (dALA) is synthesized in the mitochondria by dALA synthetase. From it are formed, successively, porphobilinogen (PBG), uroporphyrin III, coproporphyrin III, and protoporphyrin IX. Protopoyphyrin IX reenters the mitochondrion, to be acted on by ferrochelatase to produce heme. Each step in this process is catalyzed by a specific enzyme. The final product, heme, by negative feedback, represses the production, or activity, of dALA synthetase. If the amount of heme produced is inadequate due to a missing enzyme, dALA synthetase activity may be increased, leading to the production of more porphyrins. Because this enzyme system is inducible, medications that increase the cytochrome drug-metabolizing system in the liver can lead to exacerbation of the porphyrias by increasing the production of the porphyrin intermediates.
The current grouping of the porphyrias is based on the primary site of increased porphyrin production, either liver or bone marrow—the hepatic or erythropoietic porphyrias, respectively. Some include a hepatoerythropoietic category. Congenital erythropoietic porphyria (CEP), X-linked dominant protoporphyria (XLDPP), and erythropoietic protoporphyria (EPP) are the erythropoietic forms. Acute intermittent porphyria (AIP), ALA dehydratase deficiency (ADP), hereditary coproporphyria (HCP), variegate porphyria (VP), and porphyria cutanea tarda (PCT) are the hepatic forms. Hepatoerythrocytic porphyria (HEP) has been classified as either a hepatic or a hepatoerythropoietic type.
Another way to classify the porphyrias is by their symptomatology. This system divides those diseases that have acute episodes, called the acute porphyrias, and those that have skin findings, called the cutaneous porphyrias. Some conditions have both skin disease and acute episodes. The acute porphyrias are ADP, AIP, HCP, and VP. The cutaneous porphyrias are PCT, CEP, XLDPP, and EPP. VP and HCP can have both acute attacks and skin lesions. The acute attacks are induced by conditions that activate the heme biosynthesis pathway. Due to the enzymatic “blocks” as the pathway is activated, large amounts of the heme precursors (specifically, dALA and PBG) are produced by the liver and dumped into the bloodstream. These substances are neurotoxic and affect primarily the autonomic and peripheral nerves. In the cutaneous porphyrias, photosensitivity is observed. The photosensitivity is caused by the absorption of UV radiation in the Soret band (400–410 nm) by the porphyrins, primarily in the blood vessels of the upper dermis. These activated porphyrins are unstable, and as they return to a ground state, they transfer energy to oxygen, creating reactive oxygen species. These unstable oxygen species interact with biologic systems, primarily plasma and lysosomal membranes, causing tissue damage. Mediators released from mast cells and polymorphonuclear leukocytes, acting through complement and metalloproteinase, eicosanoids, or factor XII pathways, may augment tissue effects. The skin lesions are determined by the biochemical nature of the excess porphyrin. Hydrophobic protoporphyrin has more affinity to lipid membranes, specifically endothelial cells. This correlates with acute burning and purpura exhibited in EPP, as well as the prominent reduplication of the basement membranes (seen as perivascular hyaline deposits) of the upper dermal vessels from constant repair of the phototoxic damage to the endothelial cells. The more water-soluble porphyrins (uroporphyrin and coproporphyrins) diffuse into and accumulate in the dermis and along the DEJ. The resulting skin lesions, subepidermal blisters, are caused by the phototoxic damage in this region.
The porphyrias have classically been diagnosed by identifying characteristic clinical and biochemical abnormalities, typically elevated levels of porphyrins in the urine, serum, red blood cells (RBCs), or stool. Because there is some clinical overlap, biochemical testing should be performed to confirm any diagnosis of porphyria. In the acute porphyrias, patients are often asymptomatic between attacks. During attacks, porphyrin assays will be abnormal in all forms of porphyria. Between attacks, some patients with AIP may have normal porphyrin assays so genetic testing is necessary to make the diagnosis. The genetic defect and the points of the most common mutations for each gene are now known for most forms of the porphyrias. Genetic testing is now recommended in most porphyrias, except PCT and EPP. There is considerable clinical overlap in these rarer porphyrias; dual porphyrias exist, with mutations in two different heme synthesis genes; and low-level mutations causing atypical presentations are now well described. Accurate diagnosis in such cases requires genetic testing and allows for genetic counseling and prenatal diagnosis.
Porphyria Cutanea Tarda
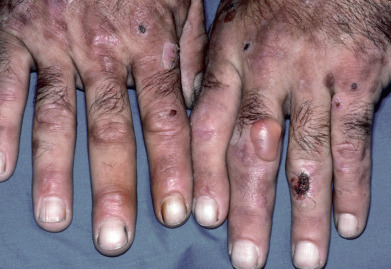
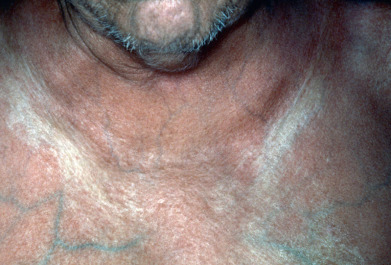
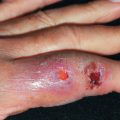
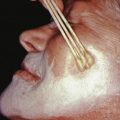
Porphyria cutanea tarda (PCT) is the most common type of porphyria. Patients with PCT present most often in midlife, averaging 45 years of age at disease onset. The disease is characterized by photosensitivity resulting in skin fragility and bullae, especially on sun-exposed parts. The dorsal hands and forearms, ears, and face are primarily affected. The bullae are noninflammatory and rupture easily to form erosions or shallow ulcers ( Fig. 26.6 ). These heal with scarring, milia, and dyspigmentation. Lesions on the legs, especially the shins and dorsal feet, occur primarily in women. There is hyperpigmentation of the skin, especially of the face, neck, and hands. Hypertrichosis of the face, especially over the cheeks and temples. The face and neck, especially in the periorbital area, may show a pink to violaceous tint. Sclerodermatous thickenings may develop on the back of the neck, in the preauricular areas, or on the thorax ( Fig. 26.7 ), fingers, and the scalp, with associated alopecia. There is a direct relationship between the levels of uroporphyrins in the urine and sclerodermatous changes.
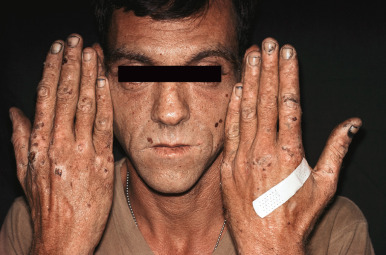
Liver disease is frequently present in patients with PCT. A history of alcoholism is common. PCT is a well-recognized cutaneous complication of hepatitis C virus (HCV) infection. All PCT patients should be screened for HCV infection especially with new therapeutics so effective for HCV. A recent report documented clearance of PCT with cure of the Hep C. Iron overload in the liver is frequently found in patients with PCT as a result of chemical or viral liver damage, or because a significant number of patients with PCT have a C282Y mutation (and a few with H63D mutation), the genetic cause of hemochromatosis ( Fig. 26.8 ). The net result of all these liver and iron metabolism abnormalities is an increase in ferritin, with hepatic iron overload. Hepatocellular carcinoma may rarely present with PCT, and PCT patients are at 3.5 times the risk of developing hepatocellular carcinoma.
PCT has been frequently associated with other diseases. It is estimated that adult-onset (type 2) diabetes mellitus occurs in 15%–20% of patients with PCT usually about a decade after the PCT diagnosis. Type 2 diabetes and the metabolic syndrome are associated with hyperferritinemia. It has been proposed that in some patients, nonalcoholic steatohepatitis (NASH) of diabetes may contribute to the development of PCT. In one patient, weight reduction led to improvement of PCT and antimalarial treatment of PCT has lead to enhanced glucose control. Moderate smoking (>10 cigarettes/day) may lead to earlier presentation of PCT by almost a decade. Numerous cases of lupus erythematosus (systemic of purely cutaneous) concomitant with PCT have been reported.
PCT can occur in patients with HIV infection. This is not only accounted for by coexistent HCV infection, which is increased in some risk groups of HIV-infected persons. Subtle porphyrin abnormalities (typically below concentrations capable of inducing disease) are found in HIV disease due to interference with normal porphyrin metabolism. Other risk factors, such as alcoholism, should be evaluated, and the existence of PCT should not be attributed to the HIV disease alone. However, effective anti-HIV therapy has led to improvement of PCT in one HIV/HCV-infected patient.
Estrogen treatment is associated with the appearance of PCT by an unknown mechanism. Before oral contraceptives were introduced, PCT cases occurred predominantly among men, but in most recent series, 60% of cases occurred in men and 40% in women. Men treated with estrogens for prostate cancer may develop PCT.
PCT is caused by a deficiency in the enzyme uroporphyrinogen decarboxylase (UROD). Several types have been described. The most common is the sporadic, nonfamilial form, which represents about 75%–80% of cases. Enzymatic activity of UROD is abnormal in the liver but normal in other tissues. This is the form associated with the cofactors previously listed. The enzyme deficiency is related to loss of enzyme activity caused by the liver damage or estrogens triggering the PCT. The enzyme UROD is inhibited by iron, so conditions that lead to iron overload in the liver (cirrhosis, alcoholism, HCV infection, type 2 diabetes, hemochromatosis) can all induce the clinical signs and symptoms of PCT. Removal of this iron in the liver may result in improvement in PCT. With remission, the enzyme activity in the liver may return to normal.
The second, or familial, type of PCT is an autosomal dominant inherited deficiency of UROD in the liver and RBCs of patients and of clinically unaffected family members. Both the activity and the concentration of the enzyme decrease by about 50%. Multiple genetic defects have been reported that produce the same phenotype. Familial PCT tends to present at an earlier age because it does not require a second insult to the liver to decrease enzyme activity further, and development of PCT before age 20 strongly suggests familial PCT.
A third form, acquired toxic PCT, is associated with acute or chronic exposure to hepatotoxins, specifically, polyhalogenated hydrocarbons such as hexachlorobenzene and dioxin. These patients have biochemical and clinical features identical to those of patients with sporadic and familial PCT.
A diagnosis of PCT can be strongly suspected on clinical grounds. A useful confirmatory test that can be performed in the office is the characteristic pink or coral-red fluorescence of a random urine specimen under Wood’s light. A 24-hour urine specimen usually contains less than 100 µg of porphyrins in a normal individual, whereas in the PCT patient it may range from 300 µg to several thousand. The ratio of uroporphyrins to coproporphyrins in PCT is typically 3 : 1 to 5 : 1, distinguishing PCT from variegate porphyria. Plasma porphyrins will also be abnormal and may be detected by peak plasma fluorescence at less than 623 nm. The diagnosis of hereditary PCT is made by demonstrating reduced UROD activity in erythrocytes. There are a few reference laboratories in the United States that have special expertise in evaluation of porphyrins and they often recommend shielding the sample from light after it is collected.
Biopsy of a blister reveals a noninflammatory subepidermal bulla with an undulating, festooned base. PAS-positive thickening of blood vessel walls in the upper and middle dermis is present. A useful and highly characteristic, but not diagnostic, feature is the presence of the so-called caterpillar bodies. These eosinophilic, elongated, wavy structures are present in the lower and middle epidermis and lie parallel to the basement membrane zone (BMZ). They stain positively with PAS and are positive for type IV collagen and laminin, suggesting they represent BMZ material present in the epidermis. DIF of involved skin shows IgG and C3 at the DEJ and in the vessel walls in a granular-linear pattern.
Initial treatment of PCT involves removal of all precipitating factors, such as alcohol, medications, and therapy for HCV and metabolic syndrome if present. This may lead to sufficient improvement so that further therapy is not required. Chemical sunscreens are of little value because they do not typically absorb radiation in the near-visible UVA range. Barrier sunscreens such as titanium dioxide and zinc oxide with nonionized zinc oxide being superior. Physical barriers such as hats and gloves should be used.
Phlebotomy is a highly effective treatment for PCT. UROD is inhibited by iron, and removal of hepatic iron may therefore lead to recovery of enzyme activity. Typically, phlebotomy of 500 mL at 2-week intervals is performed until the hemoglobin reaches 10 g/dL or the serum iron 50–60 µg/dL. Ideally, serum ferritin will become normal as well. Urinary porphyrin excretion initially increases, but gradually, 24-hour uroporphyrin levels are greatly reduced, with most patients able to achieve normal levels. This process takes several months, usually requiring a total of 6–10 phlebotomies. As the porphyrins fall, the skin lesions also involute. Initially, blistering improves, then skin fragility decreases, and finally the cutaneous sclerosis and hypertrichosis can eventually reverse. A common error in management is coadministration of oral iron supplementation during the phlebotomies to treat the anemia.
Antimalarial therapy is an alternative to phlebotomy and may be combined with phlebotomy in difficult cases. Antimalarials complex the excess porphyrins, enhancing their excretion. Full doses of antimalarials may produce a severe hepatotoxic reaction. The initial dose is 125 mg of chloroquine or 100–200 mg of hydroxychloroquine twice weekly. Improvement is gradual and parallels the reduction in porphyrins.
The duration of treatment to reach a biochemical remission is the same for phlebotomy and antimalarial therapy, about 6–7 months. This remission may last many years. If the patient relapses, these treatments can be repeated. Alternative treatments, which are rarely required, include desferrioxamine or deferasirox (iron chelation) and erythropoietin treatment. Erythropoietin may be combined with phlebotomy. PCT in renal failure may respond to erythropoietin and low-volume phlebotomy, desferrioxamine given at the end of dialysis, or renal transplantation. If HCV infection coexists, interferon alfa treatment of the HCV infection may lead to improvement of the PCT. The management of PCT associated with hemodialysis is much more difficult. High-flux, high-efficiency hemodialysis should be instituted. N -acetylcysteine, 400 mg of powder dissolved in orange juice twice daily, can be added to augment dialysis. Erythropoietin, at times at very high dose, in combination with miniphlebotomy can be used in anuric patients with PCT not controlled by other methods.
Pseudoporphyria
In certain settings, patients develop blistering and skin fragility identical to PCT, with the histologic features of PCT but with normal urine and serum porphyrins. Hypertrichosis, dyspigmentation, and cutaneous sclerosis do not occur. This pseudoporphyria is most often caused by medications, typically a nonsteroidal antiinflammatory drug (NSAID), usually naproxen. Other NSAIDs, such as nabumetone, diclofenac, and rofecoxib, as well as voriconazole, tetracycline ( Fig. 26.9 ), tolterodine, imatinib mesylate and sunitinib, metformin, finasteride, estrogen, chlorophyll, and multiple other medications, can cause a similar clinical picture. Tanning bed use can also produce pseudo-PCT. Some patients on hemodialysis develop a similar PCT-like picture. Less frequently, dialysis patients develop true PCT. In the anuric dialysis patient, true PCT and pseudo-PCT are distinguished by analysis of serum porphyrins in a laboratory knowledgeable in the normal porphyrin levels in patients undergoing hemodialysis. The treatment of pseudoporphyria is physical sun protection and discontinuance of any inciting medication. Ibuprofen is a safer alternative NSAID that usually does not cause pseudoporphyria. In medication-induced PCT, blistering resolves over several months once the medication is stopped. Skin fragility may persist for much longer. N -acetylcysteine and glutamine have been reported to improved dialysis-associated pseudo-PCT.
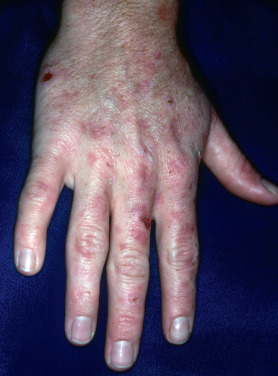
Hepatoerythropoietic Porphyria
Hepatoerythropoietic porphyria (HEP) is the homozygous form of PCT. It is caused by a homozygous or compound heterozygous deficiency of UROD, which is about 10% of normal in both the liver and the erythrocytes. The biochemical abnormalities are similar to but more marked than those in PCT, although the clinical features are similar to CEP. Dark urine is usually present from birth. In infancy, vesicles occur in sun-exposed skin, followed by sclerodermoid scarring, hypertrichosis, pigmentation, red fluorescence of the teeth under Wood’s light, and nail damage. Neurologic disease has been reported. The diagnosis of HEP is confirmed by abnormal urinary uroporphyrins (as seen in PCT), elevated erythrocyte protoporphyrins, and increased coproporphyrins in the feces. In CEP uroporphyrins are elevated in the erythrocytes, allowing differentiation from HEP. Sun protection is necessary, but often inadequate. Bone marrow transplantation, as in CEP, may be required for HEP patients.
Variegate Porphyria
Variegate porphyria (VP) is also known as mixed porphyria, South African genetic porphyria, and mixed hepatic porphyria. VP has an autosomal dominant inheritance with a high penetrance. It results from a decrease in activity of protoporphyrinogen oxidase (PPOX). Between 40% and 70% of patients with VP have skin symptoms, 27% have acute attacks, and only 14% have both acute attacks and skin symptoms. Many affected relatives have silent VP, in which there is reduced enzyme activity but no clinical lesions. Such persons should be identified and evaluated.
VP is characterized by the combination of the skin lesions of PCT and the acute GI and neurologic disease of AIP. In 50% of VP patients, skin lesions are the presenting finding. Vesicles and bullae with erosions, especially on sun-exposed areas, are the chief manifestations. In addition, hypertrichosis is seen in the temporal area, especially in women. Hyperpigmentation of sun-exposed areas is also a feature. Facial scarring and thickening of the skin may give the patient a prematurely aged appearance. The visceral attacks include hypertension, fever and paralysis of the respiratory system.
The presence of VP should be suspected in a patient when findings indicate both PCT and AIP, especially if the patient is of South African ancestry. Fecal coproporphyrins and protoporphyrins are always elevated, and during attacks, urine PBG and ALA are elevated. Normal levels of fecal protoporphyrin in adulthood predicts lack of both skin symptoms and acute attacks. Urinary coproporphyrins are increased over uroporphyrins, distinguishing VP from PCT. Urinary coproporphyrin level greater than 1000 nmol/day predicts increased risk for acute attacks and skin symptoms and indicates the need for preventive treatment to reduce porphyrins. A finding in the plasma of a unique fluorescence at 626 nm is characteristic of VP and distinguishes it from all other forms of porphyria. Lymphocyte PPOX can be measured, but because of the profound founder effect in this condition, genetic testing should be used to confirm the diagnosis.
Treatment of the skin lesions is symptomatic, because antimalarials and phlebotomy are not effective in modifying cutaneous disease in VP. Gonadotropin-releasing hormone (GnRH) analogs may prevent premenstrual attacks, and “hemin” and glucose loading can be used for acute attacks. VP patients, as well as those with other acute porphyrias (HCP and AIP), are at increased risk for hepatocellular carcinoma, and regular liver imaging should be performed after age 50. Education of patients and unaffected PPOX-deficient relatives is essential to avoid triggering medications.
Homozygous VP is a very rare autosomal recessive condition that presents in childhood with PCT-like acral blistering, vermiculate scarring of the cheeks, finger shortening, and developmental delay. Brain myelin is completely absent.
Hereditary Coproporphyria
Hereditary coproporphyria (HCP) is a rare, autosomal dominant porphyria resulting from a deficiency of coproporphyrinogen oxidase (CPO). About one third of patients are photosensitive, with blistering similar to but less severe than in VP. About 35% have acute attacks with GI and neurologic symptoms similar to those seen in AIP and VP. Fecal coproporphyrin III is always increased; urinary coproporphyrin, ALA, and PBG are increased only during attacks. Plasma fluorescence at 619 nm is seen. Mutation screening can be used to confirm the diagnosis and identify unaffected but CPO-deficient relatives. Homozygous hereditary coproporphyria, or harderoporphyria, is caused by a homozygous defect of CPO, with patients having 10% or less of normal activity. Harderoporphyrin is the natural intermediate between coproporphyrinogen and protoporphyrinogen. There is a report of HCP induced by efavirenz and liver failure in a patient who used “hydroxycut” over-the-counter weight loss therapy. Children present with photosensitivity, hypertrichosis, and hemolytic anemia, and a neonatal form was recently described. The biochemical findings in plasma, feces, and urine are identical to HCP, but more marked.
Erythropoietic Protoporphyria
Erythropoietic protoporphyria (EPP) is caused by decreased ferrochelatase (FECH) activity. There is a wild-type allele that has low function (hypomorphic) and when this is combined with a mutated allele leading to dysfunction. Therefore it is usually inherited in an autosomal dominant pattern with only one gene being truly mutated (pseudodominant). Autosomal recessive EPP from inheritance of 2 loss of function mutations is much more severe and typically requires bone marrow transplantation. FECH activity below 35% will lead to disease and most affected patients have 10%–25% of normal function. In Europe, up to 10%–15% of the population carries the low-expression (hypomorphic) allele that is only 50% as active as the wild-type enzyme.
Typically, EPP presents early in childhood (3 months to 2 years), although presentation late in adulthood can occur. The diagnosis of EPP in children is frequently delayed because of its rarity. Older children at times are referred to psychiatrists until the diagnosis is suspected.
Unique among the more common forms of porphyria is an immediate burning of the skin on sun exposure. Because the elevated protoporphyrin IX absorbs both in the Soret band and at 500–600 nm, visible light through window glass, in the operating room, or during neonatal exposure to a bili-light ( Fig. 26.10 ) may precipitate symptoms. Infants cry when exposed to sunlight. Erythema, plaquelike edema, and wheals such as those seen in solar urticaria can be seen. These lesions appear solely on sun-exposed areas. In severe cases, purpura is seen in the sun-exposed areas.
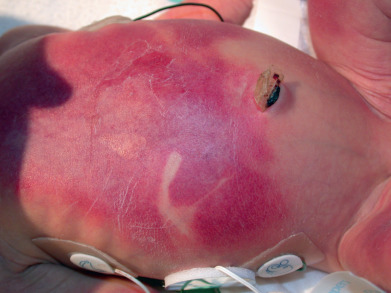
With repeated exposure, the skin develops a weather-beaten appearance. Shallow linear or elliptical scars, waxy thickening and pebbling of the skin on the nose and cheeks and over metacarpophalangeal joints, and atrophy of the rims of the ears have been described. Perioral furrow-like scars are characteristic. The dorsal hands and face of EPP patients appear much older than their chronologic age.
About 2.5% of patients with EPP have a seasonal palmar keratoderma. It is worse in the summer and resolves in winter or with occlusion of the palm by a plaster cast. The keratoderma is waxy and may cover the whole palm or may be localized to the first web space. It is sharply demarcated at the wrist and has no red border. The thickening is moderate in severity. The nails are usually unaffected but may show minimal onycholysis. Patients with nail changes all have true autosomal recessive EPP, with lower levels of erythrocyte protoporphyrin but increased levels of fecal total porphyrin compared with pseudodominant EPP patients. In fact, their erythrocyte protoporphyrin may be near normal. About 45% of patients with autosomal recessive EPP have this keratoderma.
Between 20% and 30% of EPP patients have liver complications because of excessive porphyrin deposits in hepatocytes. This can occur anywhere along the spectrum, from mild elevation of liver function tests to cirrhosis. Only 5% of patients with EPP and liver disease develop hepatic failure, or 0.5%–1% of all EPP patients. Liver transplantation may be required. It is extremely important during liver transplantation to use filters on the operating room lights. A yellow filter that only emits below 470 nm has been reported to be safe. Autosomal recessive inheritance of EPP may be a risk factor for the development of liver failure. There is currently no marker for progressive liver disease (not laboratory porphyrins or genetic defect), so all patients must be monitored. Ten percent of patients develop gallstones, often in childhood. A mild microcytic anemia is present in 25% of patients with EPP, but therapy with iron should be used only if iron deficiency is detected, because it may exacerbate symptoms. Because of sun avoidance, vitamin D deficiency can occur.
The rare syndrome of EPP appearing de novo in adults has been reported multiple times. These cases are associated with a myeloproliferative disorder or myelodysplastic syndrome. The malignant cells in the bone marrow, caused by a translocation, lose the FECH gene on chromosome 18, and the patient “acquires” a FECH deficiency. Bone marrow transplantation is associated with resolution of this form of EPP.
Histologically, there is prominent ground-glass, PAS-positive material in the upper dermis, mostly perivascularly. This material is type IV collagen. On DIF, IgG and C3 may be found perivascularly. If an acute purpuric lesion is biopsied, the features of a leukocytoclastic vasculitis may be seen.
A diagnosis of EPP can usually be suspected on clinical grounds, especially if both the acute symptoms and the chronic skin changes are found. Because protoporphyrin IX is not water soluble, urine porphyrin levels are normal. Erythrocyte protoporphyrin is elevated and can be detected by RBC fluorescence. Erythrocyte, plasma, and fecal protoporphyrin can also be assayed to confirm the diagnosis. Erythrocyte protoporphyrin levels in affected persons may range from several hundred to several thousand micrograms per 100 mL of packed RBCs (normal values, <35 µg/100 mL of packed RBCs). Plasma fluorescence shows a peak at 634 nm.
The differential diagnosis of EPP includes hydroa vacciniforme, pseudoporphyria, xeroderma pigmentosa, and solar urticaria. In infancy, before the appearance of the chronic skin changes, erythrocyte porphyrins may need to be screened to confirm the diagnosis. Once chronic changes are present, a skin biopsy will confirm the diagnosis.
The treatment of EPP patients consists of protection from exposure to sunlight with clothing and barrier sunscreens containing titanium dioxide or zinc oxide. Beta carotene, 60–180 mg/day in adults and 30–90 mg/day for children, to maintain a serum level of 600 µg/100 mL, provides some modest protection. As the child grows, the dose must be increased to maintain adequate tissue levels. Early-spring hardening with narrow-band UVB or PUVA (wavelengths below the action spectrum of the incriminated porphyrins) is being increasingly used. Preliminary trials of colestipol, 2 g daily, and oral zinc sulfate, 600 mg daily, have led to substantial increases in light tolerance in EPP patients. Alpha melanocyte stimulating hormone analogs (alfamelanotide) can help increase photoprotection by inducing melanin production. Alfamelanotide has been demonstrated to increase light tolerance in patients with EPP, in a 20-mg sustained-release form implanted every 2 months. There are reports of increased pigmentation and number of nevi.
X-Linked Dominant Protoporphyria
X-linked dominant protoporphyria (XLDPP) is caused by deletions in the dALA synthetase 2 ( ALAS2 ) gene. These mutations result in gain of function of the ALAS2 gene, with increased production of protoporphyrin. Erythrocyte protoporphyrins are elevated from this overproduction of protoporphyrin, which exceeds the capacity of the ferrochelatase to incorporate the protoporphyrin into heme, resulting in excess protoporphyrin. Patients present with symptoms identical to EPP. About 10% of patients in North America with “EPP” actually have XLDPP. More severe photosensitivity and more frequent liver disease (15%) occur in XLDPP due to higher levels of protoporphyrins, about two times higher than in EPP patients. One case of late-onset XLDPP associated with myelodysplasia has been reported. Intravenous iron therapy has improved skin symptoms.
Congenital Erythropoietic Porphyria
Congenital erythropoietic porphyria (CEP) is a very rare form of porphyria that is inherited as an autosomal recessive trait. It is caused by a homozygous defect of the enzyme uroporphyrinogen III synthase (UROS). The coinheritance of a gain-of-function mutation in ALAS2 can lead to a more severe phenotype. One family with a GATA1 mutation also developed CEP.
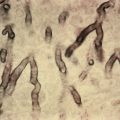
CEP presents soon after birth with the appearance of red urine (noticeable on diapers). Severe photosensitivity occurs and may result in immediate pain and burning, so that the affected child screams when exposed to the sun. The laser used in pulse oximeters may lead to skin lesions of the nail bed. Redness, swelling, and blistering occur and result in scarring of the face ( Fig. 26.11 ), dorsal hands, and scalp (with subsequent alopecia). Ectropion can occur, with subsequent corneal damage and loss of vision. Erythrodontia of both deciduous and permanent teeth is also characteristic (see Fig. 26.11 ). This phenomenon is demonstrated by the coral-red fluorescence of the teeth when exposed to Wood’s light. Mutilating scars, especially on the face, and hypertrichosis of the cheeks, with profuse eyebrows and long eyelashes, occur. Other features seen in CEP include growth retardation, hemolytic anemia, thrombocytopenia, porphyrin gallstones, osteopenia, and increased fracturing of bones.
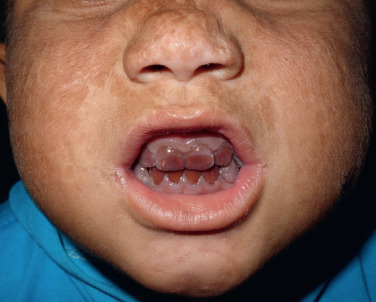
A diagnosis of CEP can be easily suspected when an infant has dark urine and is severely photosensitive. There is a direct correlation among the severity of the disease, the levels of plasma porphyrins, and the residual activity of UROS. Abnormally high amounts of uroporphyrin I and coproporphyrin I are found in urine, stool, and RBCs. There is stable red fluorescence of erythrocytes. On biopsy, a subepidermal bulla is seen, identical to that in PCT.
Treatment of CEP patients is strict avoidance of sunlight and, in some cases, splenectomy for the hemolytic anemia. Oral activated charcoal is efficacious, presumably impairing the absorption of endogenous porphyrins. Repeated transfusions of packed RBCs are given at volumes sufficient to maintain the hematocrit level at 33%, turn off the demand for heme, and reduce porphyrin production. A report of using deferasirox to induce iron deficiency also reduced symptoms. Bone marrow transplantation should be considered in severely affected children, typically those with transfusion requiring anemia or thrombocytopenia, but also those with progressive photomutilation and genotypes associated with poor outcome. Proteosome inhibitors and induced pluripotent stem cells are newer treatment opportunities.
Adult-onset CEP is extremely rare, presenting as a mild, photosensitive blistering disease resembling PCT. Usually, patients with CEP live into adulthood. Preauricular fibrosis with loss of earlobes occurs. A corticobasal syndrome resembling Parkinson disease can also occur.
Acute Intermittent Porphyria
Acute intermittent porphyria (AIP), the second most common form of porphyria after PCT, is characterized by periodic attacks of abdominal pain (up to 95% of patients), GI disturbances (up to 90% of patients), pain and paresis (50%–70%), seizures (10%–20%), and mental symptoms (40%–60%), including agitation, hallucinations, and depression. Skin lesions do not occur because the elevated porphyrin precursors are not photosensitizers. AIP is inherited as an autosomal dominant trait and is caused by a deficiency in PBG deaminase, which has 50% activity in affected persons. Only 10% of those with the genetic defect develop disease, but all may be at risk for primary liver cancer. AIP is particularly common in Scandinavia, especially Lapland. AIP usually presents after puberty in young adulthood, and women outnumber men 1.5 : 1 to 2 : 1.
Severe abdominal colic is most often the initial symptom of AIP. Patients usually have no abdominal wall rigidity, although tenderness and distention are present. Nausea, vomiting, and diarrhea or constipation accompany the abdominal pain. Peripheral neuropathy, mostly motor, is present. Severe pain in the extremities occurs. Optic atrophy, diaphragmatic weakness, respiratory paralysis, flaccid quadriplegia, facial palsy, and dysphagia are some of the many neurologic signs.
Attacks of AIP are triggered by certain medications and other conditions. These triggers frequently require increased hepatic heme synthesis (e.g., to make the cytochrome P450 enzymes required for metabolism of medications). Progesterone is one trigger, explaining the increased prevalence of AIP in women and the relationship to menses. Anticonvulsants, griseofulvin, rifampin, and sulfonamides are common drugs implicated in triggering AIP. The implicated medication list is constantly being modified as new drugs enter the market. The website of the European Porphyria Initiative is the best source for an up-to-date list of both patients and health care providers ( www.porphyria-europe.com ). Crash dieting, cigarette smoking, infections, and surgery are additional triggers.
A diagnosis of AIP is established by finding elevated levels of urinary PBG and increased dALA in the plasma and urine during attacks. During remissions, the diagnosis can be confirmed in 88% of patients by detecting elevated urinary PBG. A normal test between attacks suggests less likelihood of subsequent attacks. Erythrocyte and fecal porphyrin levels are normal. AIP must be distinguished from VP, CP, and ALA dehydratase deficiency porphyria (ALAD), an autosomal recessive condition presenting in an almost identical manner to AIP. Increased dALA in the urine is found in ALAD patients and those with lead poisoning.
No specific treatment is available for AIP. It is important for the patient to avoid such precipitating factors as a wide variety of medications, including sex steroid hormones, and to maintain adequate nutrition. Glucose loading has been used extensively and appears to be beneficial in many cases. Hematin infusions, in the form of heme arginate, result in clinical improvement and a marked decrease in ALA and PBG excretion. Early treatment may ameliorate attacks. The phenothiazines (e.g., chlorpromazine) may be helpful for pain; opiates and propoxyphene are also useful for analgesia. Because 10% of patients with AIP die of hepatoma (without the development of cirrhosis), yearly ultrasound and alpha-fetoprotein determination should be undertaken in all AIP patients over age 50.
Transient Erythroporphyria of Infancy (Purpuric Phototherapy-Induced Eruption)
Paller et al. reported seven infants exposed to 380–700 nm blue lights for the treatment of indirect hyperbilirubinemia who developed marked purpura in skin exposed to UV light. Extensive blistering and erosions occurred in one patient. Biopsies of the skin showed hemorrhage without epidermal changes in the cases associated with purpura, and a pauci-inflammatory, subepidermal bulla in the patient with blistering. The infants had all received transfusions. Elevated plasma coproporphyrins and protoporphyrins were found in the four infants examined. The pathogenesis is unknown.
Allo G, et al: Bone mineral density and vitamin D levels in erythropoietic protoporphyria. Endocrine 2013; 44: 803.
Azak A, et al: Pseudoporphyria in a hemodialysis patient successfully treated with oral glutamine. Hemodial Int 2013; 17: 466.
Badadams EL, et al: Cascade testing of primary care blood samples with hyperferritinaemia identifies subjects with iron overload and porphyria cutanea tarda. Ann Clin Biochem 2014; 51: 499.
Balwani M, et al; Porphyrias Consortium of the NIH-Sponsored Rare Diseases Clinical Research Network. Erythropoietic protoporphyria, autosomal recessive. 2012 Sep 27 [Updated 2017 Sep 7]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews [Internet]. Seattle: University of Washington, Seattle.
Balwani M, et al: Loss-of-function ferrochelatase and gain-of-function erythroid-specific 5-aminolevulinate synthase mutations causing erythropoietic protoporphyria and X-linked protoporphyria in North American patients reveal novel mutations and a high prevalence of X-linked protoporphyria. Mol Med 2013; 19: 26.
Beer K, et al: Pseudoporphyria. J Drugs Dermatol 2014; 13: 990.
Bentley DP, Meek EM: Clinical and biochemical improvement following low-dose intravenous iron therapy in a patient with erythropoietic protoporphyria. Br J Haematol 2013; 163: 277.
Berghoff AT, English JC: Imatinib mesylate–induced pseudoporphyria. J Am Acad Dermatol 2011; 63: e14.
Bishop DF, et al: Molecular expression and characterization of erythroid-specific 5-aminolevulinate synthase gain-of-function mutations causing X-linked protoporphyria. Mol Med 2013; 19: 18.
Blouin JM, et al: Therapeutic potential of proteasome inhibitors in congenital erythropoietic porphyria. Proc Natl Acad Sci USA 2013; 110: 18238.
Crawford RI, et al: Transient erythroporphyria of infancy. J Am Acad Dermatol 1996; 35: 833.
Ergen EN, et al: Is non-alcoholic steatohepatitis a predisposing factor to porphyria cutanea tarda? Photodermatol Photoimmunol Photomed 2013; 29: 106.
Erwin A, et al; Porphyrias Consortium of the NIH-Sponsored Rare Diseases Clinical Research Network. Congenital erythropoietic porphyria. 2013 Sep 12 [Updated 2016 Apr 7]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews [Internet]. Seattle: University of Washington, Seattle.
Frank J, et al: Photosensitivity in the elderly—think of late-onset protoporphyria. J Invest Dermatol 2013; 133: 1467.
García-Martín P, et al: Phototolerance induced by narrow-band UVB phototherapy in severe erythropoietic protoporphyria. Photodermatol Photoimmunol Photomed 2012; 28: 261.
Gibson GE, et al: Coexistence of lupus erythematosus and porphyria cutanea tarda in 15 patients. J Am Acad Dermatol 1998; 38: 569.
Gonzalez-Estrada A, et al: Sporadic porphyria cutanea tarda: treatment with chloroquine decreases hyperglycemia and reduces development of metabolic syndrome. Eur J Intern Med 2014; 25: e76.
Grimes R, et al: A case of hereditary coproporphyria precipitated by efavirenz. AIDS 2016; 30: 2142.
Haimowitz S, et al: Liver failure after Hydroxycut use in a patient with undiagnosed hereditary coproporphyria. J Gen Intern Med 2015; 30: 856.
Hasegawa K, et al: Neonatal-onset hereditary coproporphyria. JIMD Rep 2017; 37: 99.
Hatch MM, et al: Can curative antivirals benefit porphyria cutanea tarda in hepatitis C patients? J Eur Acad Dermatol Venereol 2017; 31: e194.
Hivnor C, et al: Cyclosporine-induced pseudoporphyria. Arch Dermatol 2003; 139: 1373.
Holme SA, et al: Seasonal palmar keratoderma in erythropoietic protoporphyria indicates autosomal recessive inheritance. J Invest Dermatol 2009; 129: 599.
Horner ME, et al: Cutaneous porphyrias part I. Int J Dermatol 2013; 52: 1464.
Jenkins SM, et al: Rash associated with phototherapy in a 2-day-old preterm male infant. NeoReviews 2016; 17: e55.
Katoulis AC, et al: Pseudoporphyria associated with nonhemodialyzed renal insufficiency, successfully treated with oral N -acetylcysteine. Case Rep Dermatol Med 2013; 2013: 271873.
Katugampola RP, et al: A management algorithm for congenital erythropoietic porphyria derived from a study of 29 cases. Br J Dermatol 2012; 167: 888.
Katugampola RP, et al: Congenital erythropoietic porphyria. Br J Dermatol 2012; 167: 901.
LaRusso J, et al: Phototherapy-induced purpuric eruption in a neonate. J Clin Aesthet Dermatol 2015; 8: 46.
Lenfestey A, et al: Metformin-induced pseudoporphyria. J Drugs Dermatol 2012; 11: 1272.
Liu LU, et al; Porphyrias Consortium of the NIH-Sponsored Rare Diseases Clinical Research Network. Familial porphyria cutanea tarda. 2013 Jun 6[Updated 2016 Sep 8]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews [Internet]. Seattle: University of Washington, Seattle.
Livideanu CB, et al: Late-onset X-linked dominant protoporphyria: an etiology of photosensitivity in the elderly. J Invest Dermatol 2013; 133: 1688.
Mungo N, et al: An unusual case of voriconazole induced pseudoporphyria. Ann Allergy Asthma Immunol 2017; 119: S24.
Oshikawa Y, et al: Photosensitivity and acute liver insufficiency in late-onset erythropoietic protoporphyria with a chromosome 18q abnormality. Case Rep Dermatol 2012; 4: 144.
Paller AS, et al: Purpuric phototherapy-induced eruption in transfused neonates. Pediatrics 1997; 100: 360.
Panton NA, et al: Iron homeostasis in porphyria cutanea tarda . J Clin Pathol 2013; 66: 160.
Pérez NO, et al: Pseudoporphyria induced by imatinib mesylate. Int J Dermatol 2014; 43: e143.
Petersen AB, et al: Zinc sulphate: a new concept of treatment of erythropoietic protoporphyria. Br J Dermatol 2012; 166: 1121.
Pham HP, et al: Therapeutic plasma exchange in a patient with erythropoietic protoporphyria status post orthotropic liver transplantation as a bridge to hematopoietic stem cell transplantation. J Clin Apher 2014; 29: 341.
Pinder VA, et al: Homozygous variegate porphyria presenting with developmental and language delay in childhood. Clin Exp Dermatol 2013; 38: 737.
Santo Domingo D, et al: Finasteride-induced pseudoporphyria. Arch Dermatol 2011; 147: 747.
Sarkany RP, et al: Acquired erythropoietic protoporphyria as a result of myelodysplasia causing loss of chromosome 18. Br J Dermatol 2006; 155: 464.
Schneider-Yin X, et al: Hepatocellular carcinoma in a variegate porphyria. Acta Derm Venereol 2010; 90: 512.
Seager MJ, et al: X-linked dominant protoporphyria. Clin Exp Dermatol 2014; 39: 35.
Sidorsky TI, et al: Development of corticobasal syndrome in a patient with congenital erythropoietic porphyria. Parkinsonism Relat Disord 2014; 20: 349.
Singal AK, Anderson KE: Variegate porphyria. 2013 Feb 14. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews [Internet]. Seattle: University of Washington, Seattle.
Singal AK, et al: Liver transplantation in the management of porphyria. Hepatology 2014; 60: 1082.
Sivaramakrishnan M, et al: Narrowband ultraviolet B phototherapy in erythropoietic protoporphyria. Br J Dermatol 2014; 170: 987.
Spelt JM, et al: Vitamin D deficiency in patients with erythropoietic protoporphyria. J Inherit Metab Dis 2010; 33: S1.
Tewari A, et al: A case of extensive hyaline deposition in facial skin caused by erythropoietic protoporphyria. Br J Dermatol 2014; 171: 412.
Thom G, et al: Leukocytoclastic vasculitis masking chronic vascular changes in previously undiagnosed erythropoietic protoporphyria. J Cutan Pathol 2013; 40: 966.
Tintle S, et al: Cutaneous porphyrias part II. Int J Dermatol 2014; 53: 3.
Tishler PV, Rosner B: Treatment of erythropoietic protoporphyria with the oral sorbent colestipol. J Am Acad Dermatol 2013; 70: 391.
Turnbull N, et al: Diclofenac-induced pseudoporphyria. Clin Exper Dermatol 2014; 39: 348.
Van Tuyll van Serooskerken AM, et al: Digenic inheritance of mutations in the coproporphyrinogen oxidase and protoporphyrinogen oxidase genes in a unique type of porphyria. J Invest Dermatol 2011; 131: 2249.
Vasconcelos P, et al: Desferrioxamine treatment of porphyria cutanea tarda in a patient with HIV and chronic renal failure. Dermatol Ther 2014; 27: 16.
Verma A, et al: Congenital erythropoietic porphyria. Br J Dermatol 2014; 171: 422.
Wahlin S, et al: Protection from phototoxic injury during surgery and endoscopy in erythropoietic protoporphyria. Liver Transpl 2008; 14: 1340.
Whatley SD, Badminton MN: Role of genetic testing in the management of patients with inherited porphyria and their families. Ann Clin Biochem 2013; 50: 204.
Willis ZI, et al: Phototoxicity, pseudoporphyria, and photo-onycholysis due to voriconazole in a pediatric patient with leukemia and invasive aspergillosis. J Pediatric Infect Dis Soc 2014; 4: e22.
Zhao CY et al: A case series of chlorophyll-induced pseudoporphyria and proposed pathogenesis. J Am Acad Dermatol 2015; 72: AB213.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


