In noninherited chronic blistering (vesicular or bullous) dermatoses, the cause of blistering is usually an autoimmune reaction, and the location where antibodies bind determines the clinical, histologic, and immunofluorescent pattern. A thorough understanding of the basement membrane zone (BMZ) structure and location of specific autoantigens and the target proteins of autoantibodies is critical. The desmoglein compensation hypothesis, based on the differential expression of desmoglein 1 and 3 at different amounts at different levels of the epidermis and mucosa, for instance, perfectly explains the observed differences in the phenotypes of pemphigus foliaceus and vulgaris. Autoantibodies that bind at the BMZ will often lead to tense bullae—but those that bind to proteins higher in that structure will often not scar, whereas antibodies against deeper parts of the BMZ will often lead to scarring and milia formation. High-frequency ultrasound has been anecdotally reported as a potential tool to determine blister location. Although the clinical and histologic findings are important, often it is the pattern of immunofluorescence that is critical in establishing the diagnosis. Usually, antibodies are circulating and can be found bound in perilesional and nonbullous lesional skin, whereas blistered skin often fails to demonstrate deposits. Biopsies of lower extremity skin should be avoided if possible, because it may be prone to false-negative results.
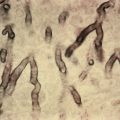
Salt-split-skin preparations are useful in determining the site of deposition of the autoantibodies. A 1-M solution of sodium chloride (NaCl) predictably splits skin at the level of the lamina lucida. Localization of immune deposits to the roof or floor of this split is diagnostically useful. On direct immunofluorescence (DIF) evaluation of nonsplit skin, the identification of n-serrated and u-serrated patterns of immunoglobulin deposition may provide the same information and may make salt-split-skin immunofluorescence unnecessary in some cases. An n-serrated pattern corresponds to a split above the basal lamina, whereas a u-serrated pattern corresponds to a sub–lamina densa split (see images on ExpertConsult). The subtle patterns are best seen in areas where the BMZ curves. Newer immunohistochemical stains such as C3d stain may also aid in the diagnosis in some cases. Immunoprecipitation, enzyme-linked immunosorbent assay (ELISA), and immunoblotting have helped to define the molecular targets of the autoantibodies and have revolutionized testing for immunobullous diseases. Data vary concerning the sensitivity and specificity of these tests, and not every test is universally available. In the setting of bullous pemphigoid, ELISA can produce apparent false-positive results at rates of 7% or higher, based on non-NC16a antibodies, as well as on anti–BP 180 antibodies that bind to the pathogenic NC16a domain but do not produce clinical disease and are not associated with positive indirect immunofluorescent findings. False-negative results also occur and are discussed later.
Transient acantholytic dermatosis (Grover disease) is an idiopathic nonimmune vesiculobullous disease that may mimic some histologic patterns of immunobullous disease, but shows no specific findings on DIF. Specific dermatoses of pregnancy are discussed under the differential diagnosis of herpes gestationis.
The outlook for immunobullous diseases has improved since the introduction of rituximab, intravenous immunoglobulins, and more targeted, autoantibody-directed therapies helping shift away from broadly toxic immunosuppressive regimens. Oral and ocular involvement often requires early aggressive therapy and a multidisciplinary approach.
Pemphigus Vulgaris
Clinical Features
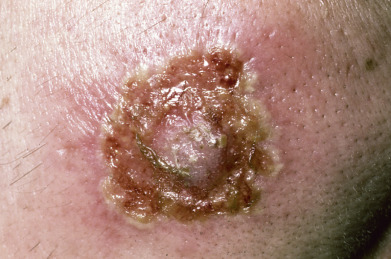
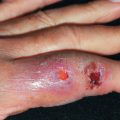
Pemphigus vulgaris (PV) is characterized by mucosal erosions and by thin-walled, relatively flaccid, easily ruptured bullae that appear on apparently normal skin and mucous membranes or on erythematous bases ( Fig. 21.1 ). The fluid in the bulla is clear at first but may become hemorrhagic or even seropurulent. The bullae rupture to form erosions. The denuded areas soon become partially or totally covered with crusts that have little or no tendency to heal. When they finally heal, lesions often leave hyperpigmented patches but no scarring.
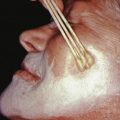
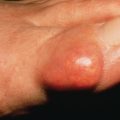
Usually, PV appears first in the mouth (60% of cases; Fig. 21.2 ) or at the site of a burn, radiation therapy, or other skin injury. Other common sites include the groin, scalp, face, neck, axillae, and genitals. Nikolsky sign is present (intact epidermis shearing away from underlying dermis, leaving a moist surface). The sign is elicited by slight pressure, twisting, or rubbing. The “bulla-spread phenomenon” (Asboe-Hansen sign) is elicited by pressure on an intact bulla, gently forcing the fluid to spread under the adjacent skin. The autoimmune bullous skin disorder intensity score (ABSIS), pemphigus vulgaris activity score (PVAS), and pemphigus disease activity index (PDAI) are scoring tools used in trials to track disease severity.
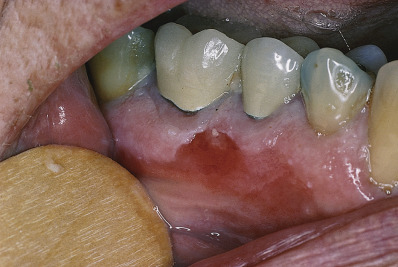
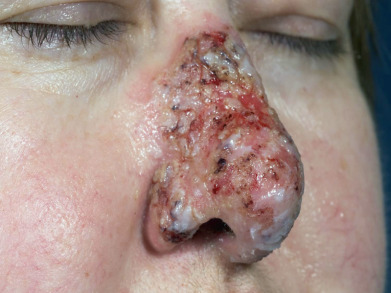
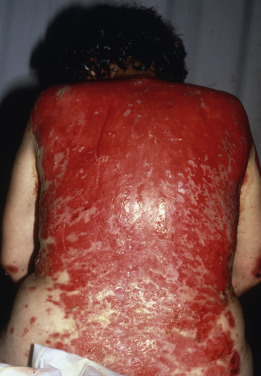
Short-lived bullae quickly rupture to involve most of the mucosa with painful erosions. The lesions extend onto the lips and form heavy, fissured crusts on the vermilion. Involvement of the throat produces hoarseness and difficulty in swallowing. The mouth odor is offensive. The esophagus may be involved, and sloughing of its entire lining in the form of a cast (esophagitis dissecans superficialis) may occur, even when the cutaneous disease appears to be well controlled because mucosa lacks desmoglein 1 and depends entirely on desmoglein 3. The conjunctiva, nasal mucosa, vagina, penis, and anus may also be involved. Chronic lesions may involve the face ( Fig. 21.3 ), scalp, or flexures. Widespread cutaneous disease ( Fig. 21.4 ) may cause death through sepsis or fluid and electrolyte imbalance.
The diagnosis is made by histology, immunofluorescence pattern of perilesional skin or plucked hairs, indirect immunofluorescence (IIF) testing of serum, or ELISA testing for anti–desmoglein 1 (Dsg1) and anti-Dsg3 autoantibodies. As in other autoimmune diseases, specific antibodies may be present in relatives of patients with pemphigus without the disease.
Epidemiology
PV occurs with equal frequency in men and women, usually in the fifth and sixth decades of life. It is rare in young persons. PV occurs more often in Jewish people and those of Mediterranean descent. Before the advent of corticosteroids, PV was frequently fatal; now, patients tend to suffer more from treatment-related complications.
Etiologic Factors
Antibodies in PV are most often directed against Dsg3. The presence of antibodies to both Dsg1 and Dsg3 correlates with mucocutaneous disease. If autoantibodies are only directed against Dsg3, mucosal lesions predominate as the mucosa lacks sufficient Dsg1 expression to compensate for the anti-Dsg3 antibody-mediated loss of keratinocyte cohesion on mucosal surfaces. Both humoral and cellular autoimmunity are important in the pathogenesis of skin lesions. Antibody alone cannot produce acantholysis, without complement or inflammatory cells. Both IgG1 and IgG4 autoantibodies to Dsg3 occur in patients with pemphigus, but some data suggest that the IgG4 antibodies are pathogenic, and patients may have circulating nonpathogenic antibodies. Plasminogen activator is associated with antibody-mediated acantholysis. Involved T cells are usually CD4 cells that secrete a T-helper type 2 (Th2)–like cytokine profile, although Th1 cells may also be involved in antibody production in chronic disease. IgG is found in both involved and clinically normal skin. C3 deposits are heavier in acantholytic areas. DIF may remain positive for years after clinical remission, and conversion to negative predicts sustained remission after withdrawal of therapy. Pemphigus may be associated with myasthenia gravis and thymoma. Recent studies suggest patients with pemphigus may have an elevated rate of ulcerative colitis. PV can affect the esophagus and cause dysphagia, and patients may be misdiagnosed with steroid-side effects initially. PV-associated interstitial lung disease is reported but very rare, and some cases may have actually had paraneoplastic pemphigus.
The PV antigen (130-kD transmembrane desmosomal glycoprotein) shows homology with the cadherin family of calcium-dependent cell adhesion molecules. With IIF, circulating antibodies can be demonstrated in 80%–90% of patients. Circulating intercellular antibodies may also be present in patients with thermal or actinic burns and in patients with drug eruptions. These antibodies are not directed against Dsg3. They do not bind to the epidermis in vivo and are often directed against ABO blood-group antigens.
Drug-induced cases are uncommon, traditionally from penicillamine or angiotensin-converting enzyme (ACE) inhibitors, though recently reports of biologic-therapy associated PV have emerged. Penicillamine treatment of rheumatoid arthritis has induced pemphigus, though more often of the foliaceous type. Almost all the reported cases have had a positive DIF, and more than half have had a positive IIF. Penicillamine and captopril may induce acantholysis in organ explant cultures in the absence of autoantibody. The doses responsible for induction of disease have ranged from 250–1500 mg/day, and the drugs were taken for an average of 13 months before the onset of pemphigus. A long list of drugs, including captopril, enalapril, penicillin, thioproline, interleukin-2 (IL-2), nifedipine, piroxicam, and rifampicin, has also been reported to induce pemphigus. Many of these contain either a sulfhydryl or an amide group. Secukinumab has been reported to induce PV in one case. Only 10%–15% of patients with drug-induced pemphigus have had oral lesions. Most disease resolves when the medication is discontinued, but some cases have persisted for many months.
Many studies have indicated a genetic predisposition to pemphigus and an association with other autoimmune diseases. A strong association exists with human leukocyte antigen (HLA)–DRB1 (with variability between specific alleles in certain ethnic/racial populations), HLA-DR4, or HLA-DR6. In addition, an HLA-DQ restriction fragment has been identified in many patients with pemphigus. A non-HLA marker “suppression of tumorigenicity 18 protein” that regulates inflammation was associated with PV in Jewish and Egyptian patients but not Europeans. The incidence in Israel is among the highest in the world, with Jewish patients, particularly Ashkenazis, at three times increased risk to Arab patients, and higher rates in women than men. HLA-G is associated with pemphigus in Jewish patients. Thus there may be a genetically inherited susceptibility to the disease. Additionally, a predisposition to develop other autoimmune diseases may occur in relatives of pemphigus patients.
Histopathology
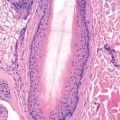
The characteristic findings of PV consist of suprabasilar acantholysis with intraepidermal blister formation. Acantholytic cells are round and show no intercellular bridges. Regeneration of the epidermis occurs and may cause the split to appear to be higher as cells regenerate beneath the cleft. At least some areas typically still demonstrate the characteristic “tombstone row” of basal keratinocytes underneath the bulla. An early intact bulla shows the most characteristic histology. Asboe-Hansen modification of the Nikolsky test may be used to extend the bulla beyond its original margin to where secondary regenerative changes have not taken place. Immunohistochemistry for IgG4 may be useful in diagnosis of PV (and PF).
In early disease, spongiosis with eosinophils may be noted in the epidermis, in the absence of acantholysis. In the setting of immunobullous disease, spongiosis with eosinophils is more likely to represent pemphigoid than pemphigus, and immunofluorescent findings readily distinguish the two. DIF demonstrates a “chicken wire” pattern of intercellular IgG in perilesional skin or plucked hairs. C3 may also be present. The staining is uniform, not granular. IIF shows a similar pattern of staining. Prozone reactions occur, so the serum should be tested at a wide range of dilutions. Positive tests may be confirmed with ELISA for the antibody.
Treatment
Pemphigus is a severe and potentially deadly disease, with patients having an increased mortality rate, and warrants aggressive immunosuppressive treatment. Large-scale, prospective, double-blinded studies are few, and the management of PV is based largely on smaller, open trials and clinical experience. A survey of 24 experienced clinicians showed that half used prednisone in doses of 1 mg/kg/day and half used higher doses. Adjuvant steroid-sparing agents were frequently employed, with almost half the respondents reporting the use of azathioprine. Because of its tolerability and simpler dosing schedule, mycophenolate mofetil (MMF) is often used in place of azathioprine. Other agents used less frequently include cyclophosphamide and methotrexate. Almost 40% of the clinicians aimed to replace prednisone with a steroid-sparing agent, whereas others were content to continue a low dose of prednisone. The survey suggests that, even among the world’s experts, there is significant variation in how this difficult disease is managed. Rituximab therapy has produced dramatic responses in some patients, and some authorities now consider rituximab appropriate first-line therapy for patients with severe disease. Intravenous immune globulin (IVIG) may also be effective, and some have combined rituximab with IVIG.
Most agents used to treat the disease are immunosuppressive, although the mechanism of action may not merely be suppression of T cells and antibody production. Methylprednisolone can directly block pemphigus antibody–induced acantholysis. It also upregulates expression of the genes encoding Dsg3 and periplakin; increases measurable levels of E-cadherin, Dsg1, and Dsg3; and interferes with phosphorylation of these adhesion molecules. Many of these effects antagonize those of pemphigus antibodies. Reversion of DIF to negative predicts sustained remission after withdrawal of medication. Plucked hairs are an alternative to skin biopsy to provide a specimen for immunofluorescence; the pilar sheath epithelium of the anagen hair typically demonstrates immunofluorescence comparable to skin.
Topical Treatment
The skin lesions are extremely painful in advanced cases. When there are extensive raw surfaces, prolonged daily baths are helpful in removing the thickened crusts and reducing the foul odor. Silver sulfadiazine (Silvadene) 1%, widely used for local therapy of burns, is an effective topical antimicrobial agent, suitable for treatment of limited disease, though broad use can lead to absorption and side effects. Silver nitrate–impregnated cotton batting, manufactured for burn units, can be used in more extensive disease. Very localized areas can be treated with silver nitrate–impregnated dressings. Painful ulcerations of the lips and mouth may benefit from topical application of a mixture of equal parts of simethicone (Maalox) and elixir of diphenhydramine hydrochloride (Benadryl) or viscous lidocaine (Xylocaine), especially before meals. The various commercial antiseptic mouthwashes are helpful in alleviating discomfort and malodor. Potent topical corticosteroids and topical tacrolimus have been successful in some patients with limited disease. The likelihood of complete remission is correlated with age of onset and initial mucosal involvement. Infection is a common complication and relates to severity of the pemphigus and the presence of diabetes mellitus.
Systemic Therapy
A common method of treatment for severe PV is to begin with doses of prednisone adequate to control the disease. High doses of prednisone (100–150 mg) are sometimes needed, but prolonged high doses are associated with significant morbidity and mortality, so adjuvant therapy should be started early. During the early phase of therapy, if prednisone at 1 mg/kg/day proves inadequate, the drug may be increased to a split dose of 1 mg/kg twice daily. As the course of corticosteroid therapy is typically longer than initially anticipated, it is good practice to begin vitamin D, calcium, weight-bearing exercise, and bisphosphonate therapy early in the course of treatment. Routine pneumocystis prophylaxis does not appear to be warranted in monotherapy, but in patients on high doses of steroids as part of combination therapy should be considered. Notably dapsone may be somewhat effective for both prophylaxis and against pemphigus. The sooner the diagnosis of PV is established and the sooner treatment is given, the more favorable the prognosis. The therapeutic effects are estimated by the number of new lesions per day and the rate of healing of new lesions. In patients with and Dsg3 antibodies, mucosal disease may still be active when cutaneous disease appears to be in remission. Pemphigus antibody titers can be performed on esophageal substrate, watching for a fall in titer. If, after 4–8 weeks of treatment, new blister formation is not suppressed, prednisone dosage may be increased to 150 mg/day. Dosage adjustments are made more frequently and aggressively in severe, progressive disease. Dividing the daily dose will usually result in greater efficacy but will also result in greater adrenal suppression. Additionally, intravenous pulse therapy with megadose corticosteroids, such as methylprednisolone (Solu-Medrol), at a dose of 1 g/day over 2–3 hours, repeated daily for 5 days, may be employed for patients unresponsive to oral doses. Untreated disease is often fatal, but the clinician should remember that, in treated patients, side effects of therapy are the most common cause of death. Adjuvant therapy to decrease steroid dependence has reduced the mortality rate.
Medication is continued until clinical disease is suppressed and pemphigus antibody disappears from the serum. Once the antibody is no longer present, a DIF test is repeated. A negative DIF is predictive of sustained remission after withdrawal of therapy.
MMF is usually chosen as a steroid-sparing agent, at a dose of 1–1.5 g twice daily. Gastrointestinal (GI) intolerance is the most common side effect, and blood counts must be monitored. If the disease does not respond, either plasmapheresis or IVIG may be added to the regimen. Azathioprine is less expensive than MMF and is often used as an alternative when cost is an overriding issue. Azathioprine is best dosed based on measurement of the patient’s thiopurine methyltransferase (TPMT) level. Most patients metabolize the drug quickly and may be underdosed if TPMT is not measured. Patients with high levels of the enzyme may require 2.5–3 mg/kg/day of azathioprine; patients with midrange levels are treated with 1–2.5 mg/kg/day. Patients deficient in TPMT may be treated with very low doses of azathioprine or with a different agent. Allopurinol interferes with metabolism of azathioprine, and increased serum levels may lead to toxicity.
Rituximab is rapidly becoming a first-line option for PV, with a recent study demonstrating efficacy as initial first-line treatment. An anti-CD20 monoclonal antibody, it leads to reduction in both CD20+ B cells, and possibly T-regulatory cells, with high response rates. There is a trend toward using rituximab early in the course of treatment if patients have significant disease, though it is still used for recalcitrant cases as well. Earlier treatment with rituximab may be associated with better clinical response. Dosing regimens include either rheumatoid arthritis dosing or lymphoma dosing, though lymphoma dosing appears to have slightly better outcomes. Patients on therapy who experience relapse tend to develop relapse as CD19+ B cells return, and when either Dsg1 or Dsg3 testing returns positive; low CD4+ T-cell count was also predictive of relapse. Human anti–chimeric antibody development can limit efficacy when they develop. Rituximab is generally well tolerated, but as with other suppressive agents, infections, including progressive multifocal leukencephalopathy, have been reported.
Patients with refractory disease may be treated with rituximab, IVIG, or cyclophosphamide, either alone or with plasmapheresis. Plasmapheresis alone is followed by rebound of antibody production, but the rebounding clone of plasma cells is sensitized to the effects of cytotoxic agents. Both daily cyclophosphamide dosing and pulse dosing schedules can be used alone or in combination with dexamethasone. Pulse dosing is usually given with mesna rescue and is associated with less bladder toxicity. Both dosing schedules should be planned early in the day, with vigorous hydration to minimize the risk of bladder toxicity. Blood counts must be monitored closely. Other risks of therapy with high doses of corticosteroids and immunosuppressants include diabetes, infection, hypertension, and cardiorespiratory disease. All these risks must be monitored, and all patients must receive gentle wound care and fluid and electrolyte management. In patients who cannot tolerate cyclophosphamide, chlorambucil has been used, but it is associated with a greater risk of hematologic malignancy. Cyclosporine and infliximab have also been used. Immunoadsorption represents a novel approach to therapy that could replace plasmapheresis. In addition to the use of IVIG as an adjuvant to conventional therapy, it has also been given as monotherapy. Onset of action is fairly rapid and may be seen within 1–2 weeks, and as an immunomodulatory and not suppressive agent with a good safety profile, it may represent a good alternative in select patients.
Immunosuppressant therapy alone has been reported as a successful treatment of patients with early, stable PV. If a contraindication to the use of corticosteroids exists, or if only limited disease is present, these may be used as single agents. In general, however, combined treatment with corticosteroids is superior in gaining early control of the disease. Dexamethasone-cyclophosphamide therapy and isolated oral cyclophosphamide are very effective options, but with high rates of adverse events. Oral cyclophosphamide was successful in 17 of 20 patients who had failed therapy with prednisone and an antimetabolite. The median time to achieve complete remission was 8.5 months, and the median duration of treatment was 17 months. Plasmapheresis was used in nine patients. Hematuria developed in five patients, and infections were noted in six. One patient developed bladder cancer 15 years after therapy.
Dapsone, nicotinamide, and tetracycline can be tried in patients with milder disease. Cyclosporine, etanercept, and infliximab have been used successfully in some patients. Intramuscular or oral gold is no longer commonly used. Gold is less effective than immunosuppressive therapy, but its advantages include lack of carcinogenicity and infertility. A minimum of 6 months is required to judge the effectiveness of gold therapy. Extracorporeal photochemotherapy has been used in a few patients. Platelet-rich plasma was reported as beneficial in a small study of seven patients with refractory oral erosions. Cutting-edge translational research into the use of specific chimeric antigen T cells to attack the pathogenic antibody-producing cells is being explored. Other monoclonal antibodies hold promise, including ofatumumab, belimumab, and veltuzumab, but there are insufficient data to recommend their use currently.
Pemphigus Vegetans
Pemphigus vegetans may present as localized plaques in the scalp or in two classic forms, the Neumann type, which generally begins and ends as typical pemphigus, and the Hallopeau type, which usually remains localized. Both types show pseudoepitheliomatous hyperplasia, and the Hallopeau type is characterized by eosinophil microabscesses within the epidermis.
Pemphigus vegetans may begin with flaccid bullae that become erosions and form fungating vegetations or papillomatous proliferations, especially in body folds or on the scalp. The tongue often shows cerebriform morphologic features early in the course of the disease. Lesions presenting as verrucous plaques on the scalp, finger, and genitalia have been reported. At times, the lesions tend to coalesce to form large patches or to arrange themselves into groups or figurate patterns.
The laboratory findings, etiologic factors, epidemiology, pathogenesis, and treatment of pemphigus vegetans are the same as those for pemphigus vulgaris. Pathogenic antibodies to desmocollins, specifically desmocollin-3 antibodies, have been reported in some cases and hypothesized to explain the different clinical features in vegetans versus vulgaris. Captopril-induced pemphigus vegetans has been reported.
Pemphigus vegetans must be differentiated from other conditions characterized by pseudoepitheliomatous hyperplasia and microabscesses, including halogenoderma, chromoblastomycosis, blastomycosis, granuloma inguinale, blastomycosis-like pyoderma, condyloma lata, and amebic granulomas. The Hallopeau type is distinguished by the presence of eosinophils, and both types by immunofluorescent findings.
Albers LN, et al: Developing biomarkers for predicting clinical relapse in pemphigus patients treated with rituximab. J Am Acad Dermatol 2017; 77: 1074.
Al-Janabi A, Greenfield S: Pemphigus vulgaris. BMJ Case Rep 2015 Oct 22; 2015.
Amber KT, et al: Determining the incidence of pneumocystis pneumonia in patients with autoimmune blistering diseases not receiving routing prophylaxis. JAMA Dermatol 2017; 153: 1137.
Atzmony L, et al: The role of adjuvant therapy in pemphigus. J Am Acad Dermatol 2015; 73: 264.
Bai YX, et al: Pemphigus vulgaris-associated interstitial lung disease. Dermatol Ther 2016; 29: 228.
Baum S, et al: Efficacy of dapsone in the treatment of pemphigus vulgaris. Dermatology 2016; 232: 578.
Baum S, et al: Epidemiological data of 290 pemphigus vulgaris patients. Eur J Dermatol 2016; 26: 382.
Cheng X, et al: High-frequency ultrasound in blistering skin diseases. J Ultrasound Med 2017; 36: 2367.
Daneshpazhooh M, et al: Immunologic prediction of relapse in patients with pemphigus vulgaris in clinical remission. J Am Acad Dermatol 2016; 74: 1160.
El-Komy MH, et al: Platelet-rich plasma for resistant oral erosions of pemphigus vulgaris. Wound Repair Regen 2015; 23: 953.
Ellebrecht CT, et al: Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science 2016; 353: 179.
Ellebrecht CT, Payne AS: Setting the target for pemphigus vulgaris therapy. JCI Insight 2017; 2: e92021.
El-Zawahry B, et al: Rituximab treatment in pemphigus vulgaris. Arch Dermatol Res 2017; Epub ahead of print.
Glauser S, et al: Diagnostic value of immunohistochemistry on formalin-fixed, paraffin-embedded skin biopsy specimens for bullous pemphigoid. Br J Dermatol 2016; 175: 988.
Hanna S, et al: Validation studies of outcome measures in pemphigus. Int J Womens Dermatol 2016; 2: 128.
Hayashida MZ, et al: Biologic therapy-induced pemphigus. An Bras Dermatol 2017; 92: 591.
Huang A, et al: Future therapies for pemphigus vulgaris. J Am Acad Dermatol 2017; 74: 746.
Inoue-Nishimoto T, et al: IgG/IgA pemphigus representing pemphigus vegetans caused by low titres of IgG and IgA antibodies to Dsg3 and IgA antibodies to desmocollin 3. J Eur Acad Dermatol Venereol 2016; 30: 1229.
Joly P, et al: First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3). Lancet 2017; 389: 2031.
Kasperkiewicz M, et al: Pemphigus. Nat Rev Dis Primers 2017; 3: 17026.
Kridin K, et al: Ulcerative colitis associated with pemphigus. Scand J Gastroenterol 2017; 52: 1360.
Kridin K, et al: Pemphigus vulgaris and pemphigus foliaceus. Acta Derm Venereol 2017; 97: 1095.
Kridin K, et al: Remarkable differences in the epidemiology of pemphigus among two ethnic populations in the same geographic region. J Am Acad Dermatol 2016; 75: 925.
Pfaltz K, et al: C3d immunohistochemistry on formalin-fixed tissue is a valuable tool in the diagnosis of bullous pemphigoid of the skin. J Cutan Pathol 2010; 37: 654.
Ruocco V, et al: Pemphigus vegetans of the folds. Clin Dermatol 2015; 33: 471.
Saruta H, et al: Two cases of pemphigus vegetans with IgG anti-desmocollin 3 antibodies. JAMA Dermatol 2013; 149: 1209.
Sinistro A, et al: The pathogenic activity of anti-desmoglein autoantibodies parallels disease severity in rituximab treated patients with pemphigus vulgaris. Eur J Dermatol 2015; 25: 578.
Svecova D: IVIG therapy in pemphigus vulgaris has corticosteroid-sparing and immunomodulatory effects. Australas J Dermatol 2016; 57: 141.
Vinay K, et al: Pemphigus vegetans presenting as a verrucous plaque on the finger. Clin Exp Dermatol 2016; 41: 316.
Wolz MM, et al: Pemphigus vegetans variant of IgA pemphigus. Am J Dermatopathol 2013; 35: e53.
Yamaguchi Y, et al: Appearance of antidesmocollin 1 autoantibodies leading to a vegetative lesion in a patient with pemphigus vulgaris. Br J Dermatol 2018; 178: 294.
Pemphigus Foliaceus
Pemphigus foliaceus (PF) is characterized by flaccid bullae and localized or generalized exfoliation. Antibodies target Dsg1. Lesions start as small, flaccid bullae that rupture almost as they appear, leading to crusting ( Figs. 21.5 and 21.6 ). Below each crust is a moist surface with a tendency to bleed. Nikolsky sign may be easily elicited by rubbing the skin. After a time, the exfoliative characteristics predominate, with few bullae. Adherent scale crusts may resemble cornflakes. A variant of pemphigus that has clinical features suggestive of dermatitis herpetiformis but has immunologic features of pemphigus has been called herpetiform pemphigus. This pustular variant can be mistaken for subcorneal pustular dermatosis, IgA pemphigus, or Sneddon-Wilkinson disease. Most of these patients represent a clinical variant of PF, with the remainder being PV patients. A few have also demonstrated desmocollin antibodies. Oral lesions are rarely seen, and then only as superficial erosive stomatitis. Several patients have been described whose clinical picture shifted from PF to PV, or vice versa, with an accompanying change in antibody profile. A rare variant of a single fixed lesion of localized pemphigus on the face or scalp has been reported.
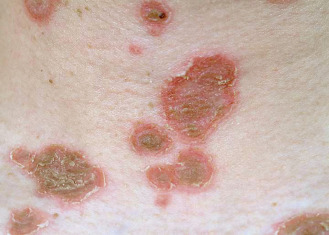
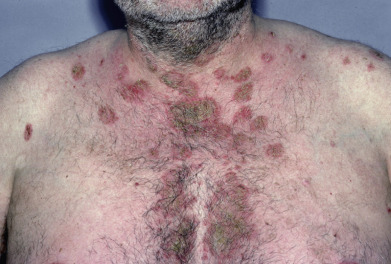
Most patients with PF are not severely ill. They complain of burning, pain, and pruritus. The lesions may persist for many years without affecting general health. PF occurs mostly in adults age 40–50 but has also been reported in children. The genders are affected equally. Prevalence of PF in people of Jewish heritage is much less than with PV. Endemic PF (fogo selvage) occurs in Brazil (see later discussion). The drugs listed under PV—penicillamine and ACE inhibitors, specifically captoprile—more frequently induce PF. Topical imiquimod and topical 5-fluorouracil have both been reported to induce PF in single case reports.
The principal histologic finding in PF consists of acantholysis in the upper epidermis, usually in the granular layer. The stratum corneum may be missing entirely or separated from the underlying epidermis. Individual elongated acantholytic cells are noted above the epidermis or clinging to the underside of the stratum corneum. DIF demonstrates intercellular IgG throughout the epidermis, although the deposits may be somewhat more prominent in the upper epidermis. IIF is positive in most patients, although prozone reactions occur and a wide range of dilutions should be tested. A sensitive and specific ELISA for detecting antibodies to Dsg1 is now available to confirm positive IIF results.
Patients with a distinct clinical picture of PF or PV may have a mix of antibodies. Western blot has shown Dsg1 in about 86% of PF patients and 25% of PV patients. ELISA has shown anti-Dsg1 antibodies in up to 71% of PF patients and 62% of PV patients. In one study, antibodies to Dsg3 were detected in 19 of 276 patients with PF and fogo selvagem who had only cutaneous disease. The antibody was capable of producing disease in laboratory animals, suggesting it was pathogenic in the PF patients. Therefore ELISA studies must always be interpreted in the context of clinical, histologic, and immunofluorescent findings. In PV, Dsg3 mediates mucosal disease, and cutaneous disease is associated with antibodies to Dsg1. A shift to predominantly Dsg1 antibodies has accompanied a clinical shift from PV to PF. Patients have also shifted from a pemphigus to a pemphigoid phenotype.
Dsg1, the antigen in PF, was first identified by immunoprecipitation consisting of polypeptides of molecular weight 260, 160, and 85 kilodaltons (kD). The 260-kD molecule is a complex of the 160-kD and 85-kD polypeptides. The PF antibody binds to a 160-kD glycoprotein extracted from normal epidermis. This glycoprotein is identical to Dsg1. The 85-kD glycoprotein is plakoglobulin, a desmosomal and adherens junction–associated molecule. Desmogleins are cadherin-type adhesion molecules found in desmosomes. The N-terminal extracellular domain of Dsg1 contains the dominant autoimmune epitopes in both PF and PV. Antibodies include both IgG1 and IgG4 subclasses. IgG4 antibodies appear to be pathogenic in most patients. In a subset of patients, IgG1 autoantibodies are pathogenic. E-cadherin autoantibodies often cross-react with Dsg1.
Treatment
Treatment of PF is similar to that for PVPF patients are generally less ill and may not need oral corticosteroid therapy. Dapsone and hydroxychloroquine may be useful, either alone in mild cases or to reduce the steroid dose level. Very mild disease may be treated with topical corticosteroids or topical calcineurin inhibitors. Nicotinamide and tetracycline may be more effective than in PV. Azathioprine, MMF, or cyclophosphamide may be needed, as in PV. As with PV, more reports are emerging of successful treatment with rituximab, with over 100 patients in the literature. Generally rituximab is effective though less dramatically than for PV, and patients with PF often require combination therapy or repeated treatments, and therefore have displayed slightly higher rates of infectious complications. The anti–IL6 receptor antibody tocilizumab, IVIG, and high-dose cyclophosphamide have been used for refractory disease. Etanercept has been used, and immunoadsorption with tryptophan-linked polyvinyl alcohol adsorbers or adsorption with plant lectins, such as wheat germ agglutinin, has been effective and holds promise as adjuvant therapy.
Herpes simplex superinfection, or S. aureus impetigo, can mimic PF and should be considered in recalcitrant or flaring cases.
Endemic Pemphigus (Fogo Selvagem)
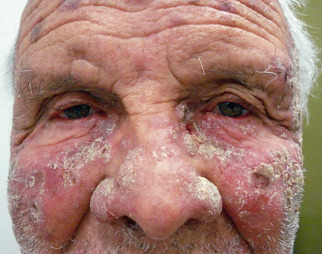
Endemic pemphigus is found in tropical regions, mostly in certain interior areas of Brazil and Colombia, but also in North Africa, including Tunisia. Fifteen percent of cases are familial. HLA-DRB1 alleles *0404, *1402, *1406, and *0102 have been identified as risk factors. The disease is common in children, adolescents, and young adults, with about one third of cases occurring before age 20 and two thirds by 40 years. The initial lesions may be flaccid bullae, but later lesions are eczematoid, psoriasiform, impetiginous, or seborrheic in appearance. The midfacial areas may be involved ( Fig. 21.7 ). Melanoderma and verrucous vegetative lesions are not unusual, and exfoliative dermatitis may occur. The mucous membranes usually are not involved. Nikolsky sign is present. The disease is often seen in those with arthropod exposure and may be initiated by an infectious agent, possibly carried by mosquitoes, sandflies, black flies, or other vectors, with those agents carrying a molecule triggering anti-Dsg1 antibodies through molecular mimicry.
Histologically and immunohistologically, fogo selvagem is identical to PF. As with PF, antibodies to desmosomal cadherins and E-cadherin may be present. The anti-Dsg1 autoantibodies cross-react with sandfly salivary LJM11 antigen. Endemic pemphigus has also been linked to the kissing bug Triatoma matogrossensis and to mercury poisoning. Peripheral blood mononuclear cells from patients produce more IL-1β than those from healthy controls. A strong Th2 bias is also observed. IgM anti-Dsg1 antibodies are common in fogo selvagem, but not in other forms of pemphigus.
A distinct subset has been described in a rural area in northeastern Colombia. This subset differs from previously described forms of endemic pemphigus and shares some immunoreactivity with paraneoplastic pemphigus. It is not, however, associated with malignant tumors. Clinically, the disease resembles Senear-Usher syndrome. A systemic form may affect internal organs and has a poorer prognosis. All patients appear to have antibodies to Dsg1. In addition, many sera react with desmoplakin I, envoplakin, and periplakin. DIF is noted in the pilosebaceous unit, adjacent neurovascular bundles and meibomian glands.
A few Brazilian sera also react with plakins. None of the Colombian patients’ sera reacted with Dsg3, but about half of Brazilian patients’ sera reacted with Dsg3. This area of Colombia is a mining region, and the population is exposed to high environmental levels of mercuric sulfides and selenides; these compounds have been found in the skin of patients with endemic pemphigus.
Pemphigus Erythematosus (Senear-Usher Syndrome)
In Senear-Usher syndrome, the early lesions are circumscribed patches of erythema and crusting that clinically resemble lupus erythematosus and are immunopathologically positive for the lupus band in 80% of patients. The lesions are erythematous and thickly crusted, bullous, or even hyperkeratotic. These are usually localized on the nose, cheeks, and ears, sites frequently affected by lupus erythematosus. In addition, crusting and impetiginous lesions appear amid bullae on the scalp, chest, and extremities. In most patients, the disease runs an indolent course. Drug-induced cases with penicillamine, captopril, propranolol, heroin, and cefuroxime have been described.
The histopathology of pemphigus erythematosus is that of PF. DIF shows IgG and complement localized in both intercellular and BMZ sites. At the dermoepidermal junction (DEJ), the deposits are continuous and granular, as in lupus. In the epidermis, they resemble those of pemphigus. Antinuclear antibody is present in low titer in 30% of patients. Patients have demonstrated anti-Dsg1 but not anti-Dsg3 autoantibodies. Additional autoantibodies may be directed against bullous pemphigoid antigen 1 (BP230) and periplakin. Patients often respond to low doses of prednisone and may respond well to topical corticosteroids. Photoprotection is essential Immunosuppressants may be needed in severe cases. Cases have been reported in association with myasthenia and thymoma. All patients should be evaluated for features of systemic lupus.
Aoki V, et al: Update on fogo selvage, an endemic form of pemphigus foliaceus. J Dermatol 2015; 42: 18.
Atzmony L, et al: Treatment of pemphigus vulgaris and pemphigus foliaceus. Am J Clin Dermatol 2014; 15: 503.
Baroni A, et al: Cefuroxime-induced pemphigus erythematosus in a young boy. Clin Exp Dermatol 2009; 34: 708.
Callander J, Ponnambath N: Pemphigus foliaceus induced by topical 5-fluorouracil. Clin Exp Dermatol 2016; 41: 443.
Caso F, et al: Refractory pemphigus foliaceus and Behçet’s disease successfully treated with tocilizumab. Immunol Res 2013; 56: 390.
de Sena Noguerira Maehara L, et al: Rituximab therapy in pemphigus foliaceus. Br J Dermatol 2015; 172: 1420.
Fernandes NC, et al: Refractory pemphigus foliaceus associated with herpesvirus infection. Rev Inst Med Trop Sao Paulo 2017; 59: e41.
Mendez-Flores S, et al: Pemphigus foliaceus with circinated plaques and neutrophil pustules. J Cutan Pathol 2016; 43: 1062.
Pritchett EN, et al: Pruritic, pink scaling plaques on the face and trunk. Pemphigus erythematosus. JAMA Dermatol 2015; 151: 1123.
Walker A, et al: Localized pemphigus foliaceus. Cutis 2017; 99: e23.
Paraneoplastic Pemphigus
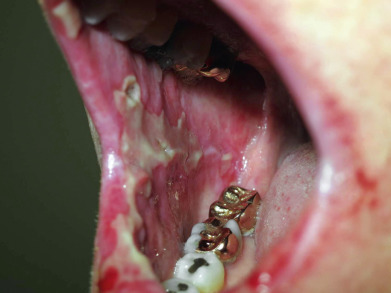
The initial description of this entity was a group of five patients with underlying neoplasms who presented with painful mucosal ulcerations and polymorphous skin lesions, which progressed to blistering eruptions. Most patients described since then have had associated neoplasms or Castleman disease. The mucosal lesions of paraneoplastic pemphigus (PNP) may appear lichenoid or may resemble Stevens-Johnson syndrome, with severe hemorrhagic crusting of the lips ( Fig. 21.8 ). The skin lesions may appear as erythematous macules, lichenoid lesions, erythema multiforme (EM)–like lesions, flaccid bullae, and erosions typical of pemphigus, or with tense, more deep-set bullae. Ocular involvement may be severe. It should be noted that all forms of pemphigus may be paraneoplastic. However, the specific disease dubbed “paraneoplastic pemphigus” has a characteristic clinical appearance as well as diagnostic immunologic findings, but it is not universally associated with a neoplasm. Many patients have devastating bronchiolitis obliterans as a manifestation of their disease.
Histologically, the lesions demonstrate epidermal acantholysis, suprabasal cleft formation, dyskeratotic keratinocytes, and vacuolar change of the basal epidermis. Biopsies that demonstrate both acantholysis and lichenoid change or individual cell necrosis should raise the suspicion of PNP. DIF reveals IgG and C3 deposition in the intercellular spaces of the epithelium. IIF shows a similar pattern in a wide range of stratified squamous epithelium and transitional epithelium (e.g., rat bladder). About 25% of cases will be negative. Immunoprecipitation is the definitive test. It reveals a complex immune response with autoantibodies directed against multiple high-molecular-weight keratinocyte proteins. Antibody targets are generally plakin family proteins at the desmosome and hemidesmosome, including desmoplakin 1 (250 kD), envoplakin (210 kD), and periplakin (190 kD), and both the major plaque protein of hemidesmosomes BPAg1 (230 kD) and plectin. Many cases also recognize an additional antigen at 170 kD. Antibodies to Dsg3, and less frequently Dsg1 and anti-α2-macroglobulin–like-1, are often present. Expanded testing has shown autoantibodies against desmocollins as well. One study suggested that antibodies to desmocollins and α2-macroglobulin–like-1 protein may be useful in making the diagnosis. Many patients have multiple antibodies. On DIF, some cases also demonstrate a linear or granular IgG and/or C3 at the BMZ. Detection of the characteristic immunologic pattern may be delayed, and tests should be repeated if the index of suspicion is high.
Whereas the dominant epitopes in PV reside in N-terminal regions of Dsg3, epitopes on Dsg3 in PNP are distributed more broadly through the extracellular domain. The N-terminal domains are still recognized more frequently than the C-terminal domains, with one study suggesting the N-terminus of envoplakin as the major epitope, particularly for patients with lichenoid inflammation and bronchiolitis obliterans. IgG subclasses in PNP are IgG1 and IgG2 dominant, contrasting with the IgG4 dominance in PV. There is a significant association in PNP with HLA-DRB1*03 allele (61.5% of those studied). In one study, eight of nine fatal PNP cases had distinctive cell surface antibodies detected in a beaded pattern by complement indirect immunofluorescence (CIIF) tests on monkey esophagus. Three long-term survivors with PNP lacked this pattern, suggesting the test may have prognostic value. One study suggested that strong cytoplasmic staining on IIF should prompt investigation for possible PNP.
A wide variety of both benign and malignant tumors are seen in these patients, and some have no identifiable neoplasm. The most common associations are hematologic malignancies, including non-Hodgkin lymphoma, chronic lymphocytic leukemia (CLL), Castleman tumor, acute myeloid leukemia (AML), and thymoma; sarcoma has also been reported. Most reported patients die of their tumor. Others have died from disease-associated bronchiolitis obliterans.
Therapy for the bullous dermatoses with prednisone and/or immunosuppressive agents should be balanced with treatment of the tumor. Severe mucosal disease, particularly ocular disease, warrants rapid, intense therapy. High dose steroids, immunoablative high-dose cyclophosphamide without stem cell rescue, cyclosporin A, plasmapheresis, immunoapheresis, and rituximab and alemtuzumab (in CLL patients), and IVIG, either alone or in combination have been successful in some cases. Occasionally the blistering disease and/or underlying disorder may be controlled, but patients may still succumb to the bronchiolitis obliterans. Even with treatment, mortality remains higher than for other immunobullous diseases.
Gong H, et al: Recurrent corneal melting in the paraneoplastic pemphigus associated with Castleman’s disease. BMC Ophthalmol 2016; 16: 106.
Hirano T, et al: Rituximab monotherapy and rituximab-containing chemotherapy were effective for paraneoplastic pemphigus accompanying follicular lymphoma, but not for subsequent bronchiolitis. J Clin Exp Hematop 2015; 55: 83.
Kartan S, et al: Paraneoplastic pemphigus and autoimmune blistering diseases associated with neoplasm. Am J Clin Dermatol 2017; 18: 105.
Namba C, et al: Paraneoplastic pemphigus associated with fatal bronchiolitis obliterans and intractable mucosal erosions. J Dermatol 2016; 43: 419.
Ohzono A, et al: Clinical and immunological findings in 104 cases of paraneoplastic pemphigus. Br J Dermatol 2015; 173: 1447.
Poot AM, et al: Direct and indirect immunofluorescence staining patterns in the diagnosis of paraneoplastic pemphigus. Br J Dermatol 2016; 174: 912.
Siddiqui S, et al: Paraneoplastic pemphigus as a presentation of acute myeloid leukemia. Hematol Oncol Stem Cell Ther 2017; 10: 155.
Wang X, et al: Extremities of the N-terminus of envoplakin and C-terminus of its linker subdomain are major epitopes of paraneoplastic pemphigus. J Dermatol Sci 2016; 84: 24.
IgA Pemphigus (Intraepidermal Neutrophilic IgA Dermatosis)
The initial description of this entity was an elderly man with chronic bullous dermatosis with unique histological and immunopathologic findings. The patient had generalized flaccid bullae, which rapidly ruptured and crusted and healed without scarring. No mucosal lesions were present. Histologically there were neutrophils linearly at the DEJ with exocytosis and some intraepidermal abscesses. DIF revealed intercellular IgA within the epidermis. No circulating antibodies were found.
Since that report, many additional patients with intraepidermal IgA deposition have been described. They have been classified as belonging to two subsets, one closely mimicking pemphigus and the second simulating subcorneal pustular dermatosis (SPD). The former starts with vesicles that become pustular within a few days, enlarge peripherally, and rupture in the center, then form a crust ( Fig. 21.9 ). Continued peripheral vesiculation may lead to a flower-like appearance. The head, neck, and trunk are frequent sites of involvement. In some patients, the condition is induced by ultraviolet (UV) A light. The second subset, SPD, presents similar to Sneddon-Wilkinson disease, with serpiginous and annular pustules. A pemphigus vegetans–like pattern has also been described. Some cases have been induced by granulocyte-macrophage colony stimulating factor (GM-CSF). Some patients have had associated malignancies, and IgA pemphigus with PNP-like clinical features has been described, showing IgA antibodies to Dsg1/3 and desmocollin 3, as well as IgG and IgA antibodies to the BMZ. Monoclonal IgA gammopathy has also been reported.