Granulatomatous reactions generally represent patterns of chronic inflammation that may take a long time to develop and a long time to respond to treatment. Granulomatous inflammation can occur in the setting of inflammatory disorders (autoimmune, autoinflammatory), medication reactions, malignancies, and infections.
Palisaded Granulomatous Dermatoses
Granuloma Annulare
Granuloma annulare (GA) is a relatively common idiopathic disorder of the dermis and subcutaneous tissue. It occurs in all races and at all ages but affects women twice as often as men. Most cases spontaneously resolve, leaving entirely normal skin, but loss of elastic tissue may occur, leaving atrophic lesions resembling middermal elastolysis or anetoderma. Long-term follow-up of at least 20 years in patients with GA reveals that lesions usually heal, and that the patients remain healthy and do not develop unusual diseases. GA may recur in a significant subset of patients, particularly with generalized lesions. GA may exhibit the isomorphic response of Koebner, may affect healed areas of herpes zoster, and may be restricted to sun-exposed areas. GA lesions will sometimes spontaneously resolve when biopsied. Case reports and retrospective reviews of associations demonstrate that GA can be a reactive condition associated with a variety of underlying disorders and medications. In most patients, however, GA is a benign, self-limited condition affecting only the skin. Variants in the GA spectrum have been subdivided with distinct names based on clinical morphologic patterns or specific histologic features, and some suggest these variants warrant consideration as separate entities.
Many morphologies of GA exist, though the vast majority of patients have localized disease, a subset have generalized disease, and children may have subcutaneous lesions. Other clinical morphologies are rare. Usually, patients exhibit primarily one clinical type during the course of their illness, except in the subcutaneous form, in which typical papular or localized GA may also occur.
Localized Granuloma Annulare
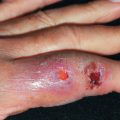
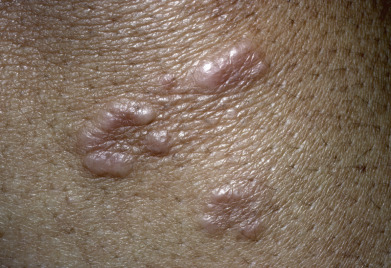
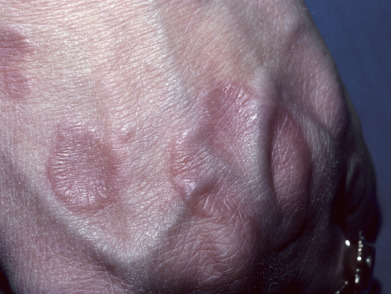
The localized form of GA tends to affect children and young to middle-aged adults. Usually, only one or a few lesions are present at any one time. Localized GA usually appears on the lateral or dorsal surfaces of the fingers or hands or dorsal feet, and occasionally on the elbows or ankles ( Figs. 31.1 to 31.3 ). Rarely, other sites, such as the eyelid or even a Becker nevus, may be affected. Lesions are erythematous to violaceous, thinly bordered plaques or papules that slowly spread peripherally while undergoing central involution, so that roughly annular lesions are formed. The overlying skin usually remains completely normal. Lesions may coalesce and sometimes form scalloped patterns or firm plaques. The lesions never ulcerate and on resolving, virtually always leave no residua. They develop slowly and often involute spontaneously. Although more than 50% of patients clear within 2 years, lesions will recur in 40%. Data regarding disease associations are limited to small series, but autoimmune thyroiditis has been described women with localized GA, though it is unclear if there is a disease-specific relationship or if both entities are simply more common in women of a particular demographic.
Generalized Granuloma Annulare
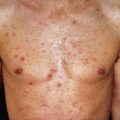
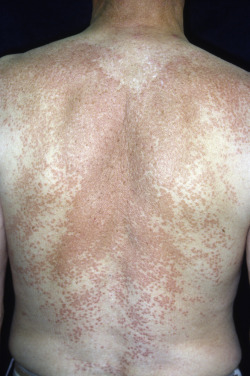
Although both disseminated and generalized GA have been described, many use the terms interchangeably. Originally, disseminated GA described patients with more than 10 lesions, and generalized GA described patients with multiple lesions involving the trunk and upper/lower extremities. Some now use disseminated if patients have predominantly papular lesions and generalized if patients have multiple annular plaques. These distinctions are based on very small case series and the prognostic, treatment, or histopathologic significance is unclear. Generalized GA affects mostly women in the fifth and sixth decades but is also a common pattern in adolescents and children. The association of generalized GA with diabetes mellitus has been questioned, although in some childhood cases diabetes and GA appeared at the same time. Similarly, dyslipidemia (elevated cholesterol, triglycerides, or low density lipoprotein (LDL) cholesterol) has been reported with generalized GA. The eruption of generalized GA presents as a diffuse but symmetric, papular or annular eruption. Lesions may number in the hundreds. Lesions favor the nape of the neck, upper trunk, and proximal extremities and rarely exceed 5 cm in diameter ( Fig. 31.4 ). The palms, soles, and eyelids may be affected. The face and genital area are usually spared. In occasional cases, sun exposure seems to be a trigger (see actinic granuloma later, under Annular Elastolytic Giant Cell Granuloma). Some patients are completely asymptomatic, whereas others complain of pruritus. Spontaneous clearing usually occurs but at variable times, and relapses may occur after clearance. The average duration is 3 to 4 years but may be as short as 4 months or longer than 10 years.
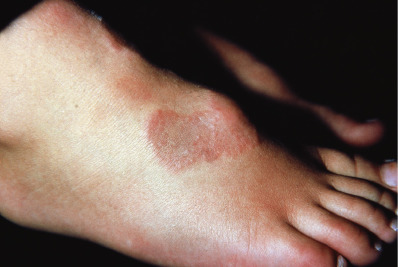
Patch-Type or Macular Granuloma Annulare
Macular GA is significantly more common in women, usually at age 30–70. Flat or only slightly palpable erythematous or red-brown lesions occur, especially on the upper medial thighs and in bathing-trunk distribution. Individual lesions average at least several centimeters in diameter but may be much larger. On careful palpation, small papules can be felt in some patients, and on stretching the skin the papules or small annular lesions can be seen.
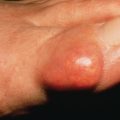
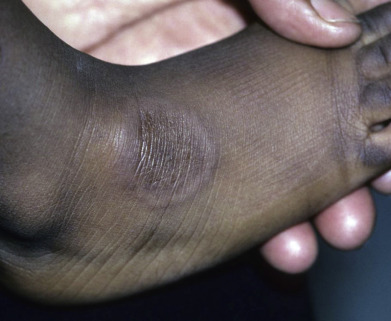
Subcutaneous Granuloma Annulare (Deep Granuloma Annulare, Pseudorheumatoid Nodule)
Subcutaneous GA is most common in children, with boys affected twice as frequently as girls. Childhood cases appear at any age from 1 year to adolescence, with one congenital case reported. Lesions tend to occur on the lower legs, especially the dorsal foot, but may also occur on the distal upper extremity or scalp. Multiple lesions are usually present. There is often a history of trauma to the affected area preceding the appearance of a lesion. Typically, lesions are skin-colored, deep dermal or subcutaneous nodules up to several centimeters in diameter ( Fig. 31.5 ). Superficial papular lesions are present in about one quarter of patients with subcutaneous GA. Lesions in general are asymptomatic and resolve over a few years. The major clinical problem occurs when the initial pathologic interpretation is “rheumatoid nodule” and an unnecessary extensive rheumatologic workup is performed. An unusual variant remains localized to the penis or scrotum, an atypical location for GA in general. Adult women without rheumatoid arthritis may develop similar lesions around the joints.
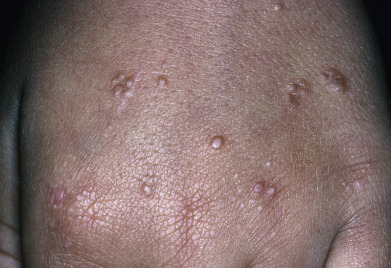
Perforating Granuloma Annulare
Perforating GA usually appears on the dorsal hands and presents as papules with a central keratotic core ( Fig. 31.6 ). This core represents transepidermal elimination of the degenerated material in the center of GA lesions and clinically can resemble a pustule. It has been suggested that in patients with atypical forms of GA it may be prudent to evaluate for underlying hematologic dyscrasia.
Palmar Granuloma Annulare/Acute-Onset Painful Acral Granuloma Annulare
This clinical variant of GA does not resemble other forms of the disease, and the diagnosis is often missed clinically. Palmar or acral GA can be chronic but is often acute. Males and females present with the sudden onset of painful lesions on the hands and feet and a scattering of lesions at other sites. The lateral, dorsal, and marginal hands and, to less extent, the feet are affected. Lesions are tender to palpation and, when present on the palms, are dusky and may vaguely resemble erythema multiforme. Patients may have associated arthralgias and diarrhea, and they feel feverish, features of a “cytokine storm.” The erythrocyte sedimentation rate (ESR) may be elevated, even above 50 mm/hr. Lesions resolve over months, at times after systemic corticosteroid or hydroxychloroquine therapy. The authors have seen one such case associated with Hodgkin disease.
Granuloma Annulare in HIV Disease
GA may occur in persons with human immunodeficiency virus (HIV) infection at all stages of disease, possibly due to cutaneous immune dysregulation. Lesions are typically papular, and generalized GA is more common (60%) than localized GA (40%). Photodistributed and perforating lesions may also occur.
Granuloma Annulare and Malignant Neoplasms
The occurrence of GA and a cancer in the same patient is rare, but it has been reported many times. Most of these patients are older. Half the cases occur in lymphoma/leukemia patients and half in those with solid tumors. The diagnosis of the neoplasm usually predates the diagnosis of GA but can precede it. In some cases, lesions are described as “atypical” in that they may be painful (see earlier), or present with less common clinical morphologic variants, such as palmar lesions.
Other Conditions Associated With Granuloma Annulare
GA has been reported after a bee sting, after waxing induced pseudofolliculitis in a patient, and after injections at a medical spa for mesotherapy or bacille Calmette-Guérin (BCG) immunization. Two groups of infectious diseases have been described as having GA-like lesions either histologically or clinically: borreliosis and tuberculosis (TB). Both Lyme disease in the United States and Borrelia infections in Europe have been described rarely as demonstrating interstitial granulomatous inflammation; clinically, however, at least in Europe, the lesions resemble morphea rather than GA. Despite laboratory evidence of infection, treatment of the patient with appropriate antibiotics may not lead to resolution of the skin lesions. A tuberculid can closely resemble disseminated GA, although histologically, caseous necrosis may be seen in the center of the granulomas. Treatment for TB leads to resolution of the skin lesions. In the appropriate patient, evaluation for TB and antituberculous treatment may be indicated. Medications can trigger interstitial granulomatous cutaneous reactions, including GA and mimickers resembling GA (see Interstitial Granulomatous Drug Reaction , later in this chapter). Tumor necrosis factor (TNF) inhibitor–induced GA has been reported, and newer anticancer agents ( BRAF inhibitors and especially checkpoint inhibitors) have been reported to cause granulomatous lesions, including GA.
Granuloma Annulare and Eye Disease
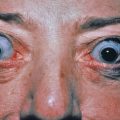
Anterior and chronic intermediate uveitis has been described in patients with localized GA. The uveitis can be unilateral or bilateral, may be mild and may respond to topical therapy, or may be aggressive, resulting in visual impairment. The frequency of uveitis in patients with GA seems to be too low to recommend that all patients with GA be screened by an ophthalmologist. However, GA patients should be questioned about visual symptoms, including reduced visual acuity. If these are present, ophthalmologic evaluation would be appropriate. Patients with granulomatous skin lesions and uveitis may also have sarcoidosis, and careful review of the histology and consideration for further screening may be warranted.
Histology
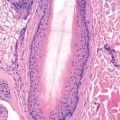
Because there are many clinical patterns of GA, skin biopsies are often performed to confirm the diagnosis. In general, two histopathologic patterns often coexist in the same patient. The classic pattern of GA is a palisading granuloma characterized by histiocytes and epithelioid cells surrounding a central zone of altered collagen. There is often mucin deposition within the foci of altered collagen. Fibrin and nuclear dust may also be present in the degenerated foci. Lesions are most often located in the upper and middle reticular dermis but may involve the deep dermis or subcutaneous tissue. At the periphery of lesions, a leukocytoclastic vasculitis may rarely be found. Immunoglobulin M (IgM) and C3 in the blood vessels of the skin lesions are found in about half of patients.
The second pattern of GA is characterized by an interstitial pattern of inflammation, which may be seen in isolation or adjacent to well-formed palisaded lesions. This histologic variant is more common than the classic palisading granuloma. A patchy dermal infiltrate of histiocytes and other mononuclear cells with occasional neutrophils is interspersed between collagen bundles. The patchy distribution within the dermis is best appreciated at scanning magnification. Interstitial mucin is often present in the affected areas. Although these features are sufficient to confirm the diagnosis of GA, further sectioning may reveal typical palisaded granulomas.
Treatment
Patients regularly report that a biopsy of the lesion will cause its involution. Because the lesions are often asymptomatic and spontaneous involution occurs, no treatment is required in many mild cases. Numerous modalities have been reported to improve GA, suggesting that no one treatment is uniformly efficacious and the “treatment of choice.” It is best to develop a therapeutic ladder for both localized and generalized cases of GA. For localized cases, the intralesional injection of triamcinolone suspension is effective and is a reasonable initial treatment. Many patients relapse within 3–7 months. Superpotent topical corticosteroids or topical calcineurin inhibitors, or imiquimod, may occasionally be effective in some patients, especially those with more macular lesions. Excimer laser, fractional photothermolysis, or photodynamic therapy can be efficacious for localized disease.
Generalized GA patients represent a major therapeutic challenge. For any treatment, 3–6 months of therapy appears necessary for efficacy or failure to be demonstrated. The two therapeutic modalities more widely reported in the literature are phototherapy and antimalarial agents. Phototherapy with psoralen ultraviolet A (PUVA) was used in initial reports, with more recent reports suggesting that narrow-band ultraviolet B (NB-UVB) may be used with benefit as well. Phototherapy should be used with caution in patients with photodistributed GA or features suggestive of possible annular elastolytic giant cell granuloma. Antimalarials have been used for decades, with initial reports describing improvement with chloroquine, and more recent data demonstrating good response rates to hydroxychloroquine, though chloroquine has successfully been used in patients who failed hydroxychloroquine.
Although systemic corticosteroids may be very effective, the high doses required and the usual immediate relapse as the steroids are tapered make this approach untenable in most situations. In addition, because dyslipidemia or metabolic syndrome may be present, systemic corticosteroids may be relatively contraindicated. Many systemic agents have been reported as effective, but few have been tested in large numbers of patients or in blinded or controlled trials. With all treatments, the GA may clear, only to recur when therapy is stopped. Antibiotics such as doxycycline; the combination of rifampin, ofloxacin, and minocycline, once monthly; pentoxiphylline, 400 mg three times daily; or high-dose nicotinamide, potassium iodide, oral calcitriol, or dapsone, 100 mg/day, can be effective. Fumaric acid esters over 1–18 months have also shown efficacy. The combination of fumaric acid esters with PUVA appears to give the highest level of response with phototherapy. Oral retinoids, especially isotretinoin, can be considered at a dose of 0.5 mg/kg or slightly more. Oral vitamin E has been used in one study of 21 patients with some improvement. For patients with severe disease, TNF inhibitors can be considered. Etanercept, infliximab, and adalimumab have all been reported to be effective. It is of interest that these medications can also cause GA. Systemic agents, such as cyclosporine, interferon (IFN) gamma, and hydroxyurea, have been reported to be effective in small series of patients. The potential toxicity of these medications limits their use to patients with significant GA.
Adams DC, Hogan DJ: Improvement of chronic generalized granuloma annulare with isotretinoin. Arch Dermatol 2002; 138: 1518.
Badavanis G, et al: Successful treatment of granuloma annulare with imiquimod cream 5%. Acta Derm Venereol 2005; 85: 547.
Balin SJ, et al: Myelodysplastic syndrome presenting as generalized granulomatous dermatitis. Arch Dermatol 2011; 147: 331.
Barzilai A, et al: Pseudorheumatoid nodules in adults: a juxta-articular form of nodular granuloma annulare. Am J Dermatopathol 2005; 27: 1.
Baskan EB, et al: A case of granuloma annulare in a child following tetanus and diphtheria toxoid vaccination. J Eur Acad Dermatol Venereol 2005; 19: 639.
Brey NV, et al: Acute-onset, painful acral granuloma annulare. Arch Dermatol 2006; 142: 49.
Brey NV, et al: Association of inflammatory eye disease with granuloma annulare? Arch Dermatol 2008; 144: 803.
Cunningham L, et al: The efficacy of PUVA and narrowband UVB phototherapy in the management of generalised granuloma annulare. J Dermatolog Treat 2016; 27: 136.
Czarnecki DB, et al: The response of generalized granuloma annulare to dapsone. Acta Dermatol Venereol (Stockh) 1986; 66: 82.
Dahl MV: Testing lipid levels in granuloma annulare. Arch Dermatol 2012; 148: 1136.
Garg S, Baveja S: Generalized granuloma annulare treated with monthly rifampicin, ofloxacin and minocycline combination therapy. Indian J Dermatol 2013; 58: 197.
Garg S, Baveja S: Monthly rifampicin, ofloxacin, and minocycline therapy for generalized and localized granuloma annulare. Indian J Dermatol Venereol Leprol 2015; 81: 35.
Giunta A, et al: Granuloma annulare. G Ital Dermatol Venereol 2017; 152: 193.
Grewal SK, et al: Antimalarial therapy for granuloma annulare. J Am Acad Dermatol 2017; 76: 765.
Gualco F, et al: Interstitial granuloma annulare and borreliosis. J Eur Acad Dermatol Venereol 2007; 21: 1117.
Karsai S, et al: Fractional photothermolysis for the treatment of granuloma annulare. Lasers Surg Med 2008; 40: 319.
Keimig EL: Granuloma annulare. Dermatol Clin 2015; 33: 315.
Klein A, et al: Off-label use of fumarate therapy for granulomatous and inflammatory skin diseases other than psoriasis vulgaris. J Eur Acad Dermatol Venereol 2012; 26: 1400.
Lee SB, et al: Vemurafenib-induced granuloma annulare. J Dtsch Dermatol Ges 2016; 14: 305.
Levin NA, et al: Resolution of patch-type granuloma annulare lesions after biopsy. J Am Acad Dermatol 2002; 46: 426.
Levy J, et al: Granuloma annulare as an isotopic response to zoster. J Cutan Med Surg 2014; 18: 413.
Lukacs J, et al: Treatment of generalized granuloma annulare. J Eur Acad Dermatol Venereol 2015; 29: 1467.
Marcus DV, et al: Granuloma annulare treated with rifampin, ofloxacin, and minocycline combination therapy. Arch Dermatol 2009; 145: 787.
Min MS, et al: Treatment of recalcitrant granuloma annulare with adalimumab. J Am Acad Dermatol 2016; 74: 127.
Nebesio CL, et al: Lack of an association between granuloma annulare and type 2 diabetes mellitus. Br J Dermatol 2002; 146: 122.
Patrizi A, et al: Childhood granuloma annulare. G Ital Dermatol Venereol 2014; 149: 663.
Pavlovsky M, et al: NB-UVB phototherapy for generalized granuloma annulare. Dermatol Ther 2016; 29: 152.
Piette EW, Rosenbach M: Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol 2016; 75: 457.
Piette EW, Rosenbach M: Granuloma annulare: pathogenesis, disease associations and triggers, and therapeutic options. J Am Acad Dermatol 2016; 75: 467.
Poppe H, et al: Treatment of disseminated GA with oral vitamin E. Dermatology 2013; 227: 83.
Simpson B, et al: Triple antibiotic combination therapy may improve but not resolve granuloma annulare. Dermatol Ther 2014; 27: 343.
Thornsberry LA, English JC 3rd: Etiology, diagnosis and therapeutic management of granuloma annulare. Am J Clin Dermatol 2013; 14: 279.
Toro JR, et al: Granuloma annulare and human immunodeficiency virus infection. Arch Dermatol 1999; 135: 1341.
Verne SH, et al: Laser treatment of granuloma annulare. Int J Dermatol 2016; 55: 376.
Weinberg JM, et al: Granuloma annulare restricted to Becker’s nevus. Br J Dermatol 2004; 151: 245.
Wollina U, Langner D: Treatment of disseminated granuloma annulare recalcitrant to topical therapy. J Eur Acad Dermatol Venereol 2012; 26: 1319.
Wu W, et al: Dyslipidemia in granuloma annulare. Arch Dermatol 2012; 148: 1131.
Zhong W, et al: Perforating granuloma annulare. J Eur Acad Dermatol Venereol 2016; 30: 1246.
Annular Elastolytic Giant Cell Granuloma (Meischer), Annular Elastolytic Granuloma, and Actinic Granuloma (O’brien)
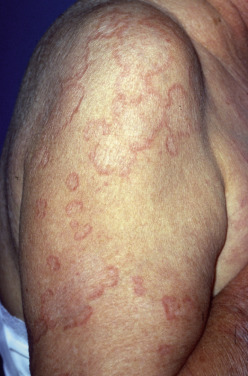
Annular elastolytic giant cell granuloma (AEGCG) and actinic granuloma are unified by their histopathologic appearance. Annular elastolytic granuloma (AEG) has been proposed as an alternative term to describe this spectrum of cases. Perhaps some cases called facial annular sarcoidosis and non–diabetes-associated necrobiosis lipoidica (NL) of the face can been included in this category. It is currently unclear whether these simply represent variants of GA, occurring most frequently on sun-damaged skin, or are distinct diseases.
Two patterns of AEGCG have been reported. The first is a single, asymptomatic, atrophic-appearing, yellow, thin plaque on the forehead (Meischer granuloma). Fine wrinkling and loss of elasticity characterize the skin within the ring. Clinically, this pattern resembles facial NL more than GA. The second variant consists of multiple extensor upper extremity and sometimes trunk lesions, occurs more frequently in women, and favors sun-exposed areas. In these cases, the lesions have an active erythematous-to-yellow/orange border with central clearing or whiter centers ( Fig. 31.7 ). A papular variant has been described. Although the vast majority of cases occur in adults, children and even an infant have been affected. Most patients are otherwise well, though reports exist of rare associations with temporal arteritis. However, AEGCG has been described in association with acute myelogenous leukemia (which resolved with remission and recurred with relapse of the leukemia) and pleomorphic cutaneous T-cell lymphoma. At times, as in GA, the lesions of AEGCG may heal with loss of elastic tissue and clinical features of skin laxity and anetoderma. The condition is chronic.
Actinic granuloma, as described by O’Brien, may represent the same disorder as AEGCG. It presents as papules and plaques on sun-exposed skin. Lesions are frequently numerous and may coalesce to cover much of the exposed skin. A history of onset after significant sun exposure and the distribution on physical examination should lead to suspicion of the diagnosis. A few lesions may occur on sun-protected sites or may spill over from affected areas to more photoprotected sites. Rarely, open comedones, scarring, and milia formation may be present clinically. Actinic granuloma may be associated with transepidermal elimination of damaged connective tissue or loss of elastic tissue surrounding the follicular ostia, leading to a Favre-Racouchot–like appearance. Repigmentation of gray hairs and improvement in solar damage within the center of lesions, have been described. This condition affects older adults (usually over age 50) and can be intensely pruritic. Actinic granuloma is not associated with diabetes mellitus, but in numerous reports, it occurred in patients with temporal arteritis. It is speculated that the vasculitis is also caused by actinic injury to the connective tissue surrounding the temporal artery. Notably both GA lesions and temporal arteritis have been reported following varicella-zoster virus infection as well. Conjunctival involvement has been reported.
Histologically, all these conditions show a characteristic histology. The dermal infiltrate of macrophages is largely interstitial, and well-formed palisaded granulomas are uncommon. Multinucleated giant cells, often quite large, are numerous. Mucin is scant or lacking. The macrophages characteristically contain fragments of actinically damaged elastic tissue (elastophagocytosis). When this typical histology is seen in concert with the classic clinical features previously noted, it may be reasonable to make these specific diagnoses. These conditions cannot, however, be diagnosed on clinical or histologic grounds alone. Some cases with the clinical features of AEGCG or actinic granuloma will show histology more characteristic of typical GA or even sarcoidosis, suggesting a spectrum of both clinical and histologic features in these patients. Other granulomatous disorders can also demonstrate elastophagocytosis on histology as well.
Treatment of AEGCG (annular elastolytic granuloma) and actinic granuloma has been difficult. Aggressive sun protection should be encouraged for patients with lesions primarily on sun-exposed skin. Topical and intralesional corticosteroids and topical calcineurin inhibitors can be used for individual lesions. Many patients respond to systemic corticosteroids, but relapse immediately when the steroids are tapered or discontinued. Oral antimalarials are at times effective, as is dapsone. Insulin improved diabetic control and the actinic granuloma in one patient. Other anecdotal treatments include oral retinoids, tetracycline-class antibiotics, fumaric acid, PUVA, pentoxiphylline, tranilast, cyclosporine, and methotrexate. TNF inhibitors have been beneficial for recalcitrant cases.
Neutrophilic sebaceous adenitis presents with asymptomatic annular plaques on the face of men more than women. Fewer than 10 cases have been reported. It may be photosensitive. The condition resolves spontaneously after weeks to months without scarring. Histologically, in early lesions there is a neutrophilic, multifocal infiltrate around sebaceous glands with necrosis of some sebocytes. In later lesions the inflammation is primarily lymphohistiocytic. In addition to granuloma annulare/annular elastolytic giant cell granuloma, tinea facei, pemphigus foliaceus, a gyrate erythema, and lupus erythematosus are in the clinical differential diagnosis.
Berliner JG, et al: The sarcoidal variant of annular elastolytic granuloma. J Cutan Pathol 2013; 40:917.
Can B, et al: Successful treatment of annular elastolytic giant cell granuloma with hydroxychloroquine. Int J Dermatol 2013; 52: 509.
De Oliveira FL, et al: Hybrid clinical and histopathological pattern in annular lesions. Case Rep Dermatol Med 2012; 2012: 102915.
Errichetti E, et al: Annular elastolytic giant cell granuloma treated with topical pimecrolimus. Indian J Dermatol Venereol Leprol 2014; 80: 475.
Fernandez-Florez A, et al: Repigmentation of gray hairs in lesions of annular elastolytic giant cell granuloma. Cutis 2015; 96: E19.
Goldminz AM, Gottlieb AB: Noninfectious granulomatous dermatitides. Part 2 of 3. Semin Cutan Med Surg 2013; 32:e1.
Haimovic A, et al: Annular elastolytic giant cell granuloma successfully treated with adalimumab subsequently complicated by drug-induced lupus. J Drugs Dermatol 2017; 16: 169.
Monserrat Garcia MT, et al: Annular elastolytic giant cell granuloma in sun-protected sites responds to dapsone. Actas Dermosifiliogr 2016; 107: 531.
Nanbu A, et al: Annular elastolytic giant cell granuloma successfully treated with minocycline. Acta Derm Venereol 2015; 95: 756.
Panzarelli A, et al: Annular elastolytic giant cell granuloma and temporal arteritis following herpes zoster. Skinmed 2015; 13: 321.
Ruocco E, et al: Annular elastolytic giant cell granuloma and temporal arteritis following herpes zoster. Skinmed 2015; 13: 267.
Interstitial Granulomatous Drug Reaction
Interstitial granulomatous drug reaction (IGDR) is an uncommon but increasingly recognized pattern of adverse reactions to medication. Cases reported as GA induced by a medication often have an “interstitial” pattern and lack mucin, and some of the same medications cause both IGDR and “GA,” so medication-induced GA and IGDR are considered together here. Some authors have suggested the unifying term reactive granulomatous dermatitis, to include the spectrum of disorders ranging from palisaded neutrophilic and granulomatous dermatitis, to interstitial dermatitis, to interstitial granulomatous drug reaction, as each of those entities requires similar workup and has a similar list of potential drug-inducing culprits. Most patients with IGDR have been taking the medication for months to years, though the reaction may occur more quickly. A wide variety of medications have been implicated, including calcium channel blockers (most common cause reported), lipid-lowering agents, angiotensin-converting enzyme (ACE) inhibitors, diuretics, nonsteroidal antiinflammatory drugs (NSAIDs), antihistamines, anticonvulsants including gabapentin, antidepressants, allopurinol and febuxostat, darifenacin, sorafenib, ganciclovir, trastuzumab, strontium ranelate, sennoside (common over-the-counter laxative), Chinese herbs, and even soy. Immunomodulatory medications, including thalidomide, leflunomide, lenalidomide, anakinra, IFNs alpha and beta, and TNF inhibitors have been implicated in causing IGDR in many cases. Rarely, drug-induced hypersensitivity syndrome (DIHS/DRESS) will display the histology of IGDR.
Clinically, the lesions are erythematous-to-mauve or violaceous annular thing plaques or patches with an indurated border and sometimes a tendency to central lightening. Lesions favor the creases (groin, axillae, popliteal fossae) but may also affect the trunk, proximal extremities, and rarely the palms and soles. Lesions may uncommonly be photodistributed, affecting the face and dorsal extensor forearm and hands. Pruritus is minimal or absent. Mucous membranes are spared. Histologically, there is a diffuse dermal infiltrate that is perivascular but has a prominent interstitial component. The inflammatory infiltrate is centered in the lower dermis; it contains neutrophils, eosinophils, histiocytes, and small multinucleated giant cells (sometimes just two or three nuclei). Degenerated collagen bundles may be surrounded by histiocytes, neutrophils, and eosinophils, resembling “Churg-Strauss” granulomas, and mucin is usually scant or absent. Unique features that should suggest IGDR over GA include an interface component and “atypical” lymphocytes in the infiltrate. The histologic differential diagnosis includes interstitial granulomatous dermatitis, palisaded neutrophilic and granulomatous dermatitis, and interstitial GA. Lesions resolve over months once the offending agent is stopped.
Cassone G, Tumiati B: Granuloma annulare as a possible new adverse effect of topiramate. Int J Dermatol 2014; 53: 259.
Chen YC, et al: Interstitial granulomatous drug reaction presenting as erythroderma: remission after discontinuation of enalapril maleate. Br J Dermatol 2008; 158: 1143.
Cornillier H, et al: Interstitial granulomatous dermatitis occurring in a patient with SAPHO one month after starting leflunomide, and disappearing with ustekinumab. Eur J Dermatol 2016;26:614-5.
Coutinho I, et al: Interstitial granulomatous dermatitis. Am J Dermatopathol 2015; 37: 614.
Deng A, et al: Interstitial granulomatous dermatitis associated with the use of tumor necrosis factor alpha inhibitors. Arch Dermatol 2006; 142: 198.
Fernando SL, et al: Drug-induced hypersensitivity syndrome with superficial granulomatous dermatitis-a novel finding. Am J Dermatopathol 2009; 31: 611.
Georgesen C, et al: Interstitial granulomatous dermatitis associated with gabapentin. Dermatitis 2014; 25: 374.
Goldminz AM, Gottlieb AB: Noninfectious granulomatous dermatitides. Part 3 of 3. Semin Cutan Med Surg 2013; 32: e7.
Hernandez N, et al: Generalized erythematous-violaceous plaques in a patient with a history of dyslipidemia. Int J Dermatol 2013; 52: 393.
Kim MS, et al: Allopurinol-induced DRESS syndrome with a histologic pattern consistent with interstitial granulomatous drug reaction. Am J Dermatopathol 2014; 36: 193.
Laura A, et al: Interstitial granulomatous drug reaction due to febuxostat. Indian J Dermatol Venereol Leprol 2014; 80: 182.
Magro CM, et al: The interstitial granulomatous drug reaction. J Cutan Pathol 1998; 25: 72.
Ratnarathorn M, et al: Disseminated granuloma annulare: a cutaneous adverse effect of anti-TNF agents. Indian J Dermatol 2011; 56: 752.
Rosenbach M, English JC 3rd: Reactive granulomatous dermatitis. Dermatol Clin 2015; 33: 373.
Tan ES, et al: Interstitial granulomatous drug reaction induced by quetiapine. Clin Exp Dermatol 2016; 41: 210.
Granuloma Multiforme (Leiker)
Granuloma multiforme (GM) is seen most frequently in central Africa, where it is a common disorder, and rarely elsewhere. It affects adults over age 40 and is more common in women. Lesions are most frequently found on the upper trunk and arms and in sun-exposed areas. GM begins as small papules that evolve within 1 year into round or oval plaques up to 15 cm in diameter. The active edge of lesions may be elevated to as much as 4 mm in height and the center slightly depressed and hypopigmented. Pruritus can occur, and coalescing lesions may form unusual polycyclic shapes. The course is chronic. GM is, most importantly, separated from tuberculoid leprosy. Histologically, GM resembles GA, but multinucleated giant cells are prominent. Giant cells typically contain phagocytosed connective tissue, and elastic tissue is decreased in the areas affected by the granulomas. GM shares many features with AEGCG and actinic granuloma, or GA of sun-exposed skin, and in fact may be considered identical to these disorders.
Kumari R, et al: Granuloma multiforme. Indian J Dermatol Venereol Leprol 2009; 75: 296.
Necrobiotic Xanthogranuloma
Necrobiotic xanthogranuloma (NXG) is a rare multisystem disease with prominent skin findings. The cause is unknown, though there is a strong association with paraproteinemia. Some consider it to exist along a spectrum of “adult orbital xanthogranulomatous disease” (AOXGD), which also includes adult-onset xanthogranuloma (AOX), and adult-onset asthma with periocular xanthogranuloma (AAPOX). NXG is gradually progressive, affecting men and women equally, and beginning on average at about age 50 (range 25–80 or older). The most common site affected is the periorbital area (>80% of patients). Multicentric involvement is typical. Lesions may be localized or initially present in scars. The characteristic skin lesions are violaceous plaques and nodules with a prominent yellow (xanthomatous) component. Periorbitally, early lesions may be mistaken for xanthelasma, but NXG lesions are more deep, firm, and indurated and may extend into the orbit and ulcerate. The trunk and proximal extremities may have violaceous to orange-red plaques with an active red border and an atrophic center with superficial telangiectasias ( Fig. 31.8 ). These plaques may grow to 25 cm in diameter. The skin lesions ulcerate in 50% of cases, leading to atrophic scarring. Acral nodules may also occur, some localized solely to the subcutaneous tissue. Extensive lesions limited to the vulvar region have been described. Extracutaneous involvement most often affects the eyes. Patients may complain of burning, itching, or pain around or in the eyes. Diplopia and inflammation in various compartments of the eye can occur, including conjunctivitis, keratitis, scleritis, uveitis, iritis, ectropion, or proptosis. Ulceration and scarring of the plaques and distortion of the eye may lead to visual occlusion. Blindness may result. Lymphadenopathy, hepatosplenomegaly, and mucosal, myocardial, and pulmonary lesions may occur. As with other granulomatous processes, the granulomas may be metabolically active and produce 1α-hydroxylase, leading to increased 1,25-dihydroxyvitamin D and hypercalcemia. Most importantly, there is a monoclonal IgG (usually κ) paraproteinemia in 80% of cases, though other paraproteins have been reported including rarely an IgA paraproteinemia. Thrombocytopenia, neutrophilia, neutropenia, and eosinophilia may be present. The bone marrow may show leukopenia, plasmacytosis (25%–50% of patients), or frank myeloma (10%–20%). In some patients, a myelodysplastic syndrome may be present or may develop (chronic lymphocytic lymphoma, Hodgkin or non-Hodgkin lymphoma). Many patients are diagnosed with “monoclonal gammopathy of unknown significance” (MGUS), as 5%–10% of patients over 70 may display a paraprotein. It is important to confer with patients’ hematologists about the significance of the NXG and relationship between the disease and myeloma. The NXG predates the development of the myeloma or myelodysplastic syndrome by an average of 2.5–5 years, but has been reported as long as 20 years preceding an eventual diagnosis of myeloma.
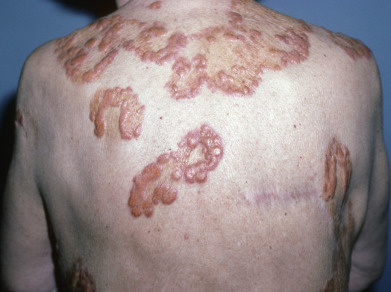
Histologically, there are extensive zones of degenerated collagen surrounded by palisaded macrophages. These macrophages are of various forms: foamy, Touton cells, epithelioid, and giant cells, sometimes with more than 50 nuclei. Peripheral foamy cytoplasm is common. Atypical multinucleated giant cells with multiple nuclei clustered at one end of the cell (polarized nuclei) are seen in 80% or more of cases. The process extends into the fat, obliterating fat lobules. Cholesterol clefts and extracellular lipid deposits may be prominent, but not universally present and are not required for the diagnosis. Within this process is a perivascular and interstitial infiltrate of lymphocytes and plasma cells. Lymphoid follicles are present. In the skin, the lymphoid aggregates are polytypic. The histologic differential diagnosis includes NL and other histiocytoses. NXG has more atypical and Touton giant cells, lymphoid nodules, and cholesterol clefts. As in plane xanthomas seen with paraproteinemia, the associated monoclonal gammopathy of undetermined significance (MGUS) appears to enhance the intracellular accumulation of cholesterol within the macrophages/histiocytes.
The treatment is usually directed at the paraprotein or underlying malignancy. Treatment of the malignancy may lead to resolution of the NXG lesions. Other treatments have included intralesional or topical corticosteroids, laser therapy, systemic corticosteroids, topical mechlorethamine, hydroxychloroquine, dapsone, IFN alpha, systemic chemotherapeutic agents (e.g., chlorambucil, cyclophosphamide, melphalan, fludarabine), plasmapheresis, or local radiation therapy (for eye lesions). Numerous reports have documented response to high-dose intravenous immunoglobin (which may in fact be treating the underlying paraproteinemia). In addition, rituximab, extracorporeal photophoresis, and thalidomide have induced remissions. Infliximab, azathioprine, and methotrexate have also been used. Simple excision is an option, but lesions may recur.
Abdul-Hay M: Immunomodulatory drugs for the treatment of periorbital necrobiotic xanthogranuloma. Clin Adv Hematol Oncol 2013; 11: 680.
Ali FR, Young HS: Ophthalmological considerations in necrobiotic xanthogranuloma. Clin Exp Dermatol 2016; 41: 563.
Bhari N, et al: Necrobiotic xanthogranuloma with multiple myeloma. Clin Exp Dermatol 2015; 40: 811.
DeLuca IJ, Grossman ME : Vulvar necrobiotic xanthogranuloma. J Am Acad Dermatol 2014; 71: e247.
Hallerman C, et al: Successful treatment of necrobiotic xanthogranuloma with intravenous immunoglobin. Arch Dermatol 2010; 146: 957.
Higgins LS, et al: Clinical features and treatment outcomes of patients with necrobiotic xanthogranuloma associated with monoclonal gammopathies. Clin Lymphoma Myeloma Leuk 2016; 16: 447.
Koch PS, et al: Erythematous papules, plaques and nodular lesions on the trunk and within preexisting scars. JAMA Dermatol 2013; 149: 1103.
Lam K, et al: Bilateral necrobiotic xanthogranuloma of the eyelids followed by a diagnosis of multiple myeloma 20 years later. Ophthal Plast Reconstr Surg 2013; 29: e118.
Liszewski W, et al: Treatment of refractory necrobiotic xanthogranulomas with extracorporeal photopheresis and intravenous immunoglobulin. Dermatol Ther 2014; 27: 268.
Miguel D, et al: Treatment of necrobiotic xanthogranuloma—a systematic review. J Eur Acad Dermatol Venereol 2017; 31: 221.
Minami-Hori M, et al: Adult orbital xanthogranulomatous disease. Clin Exp Dermatol 2011; 36: 628.
Nambudiri VE, et al: Successful multimodality treatment of recalcitrant necrobiotic xanthogranuloma using electron beam radiation and intravenous immunoglobulin. Clin Exp Dermatol 2016; 41: 179.
Rodriguez O, et al: Necrobiotic xanthogranuloma treated with topical nitrogen mustard. JAMA Dermatol 2016; 152: 589.
Rubinstein A, et al: Successful treatment of necrobiotic xanthogranuloma with intravenous immunoglobulin. J Cutan Med Surg 2013; 17: 347.
Sfeir JG, et al: Hypercalcemia in necrobiotic xanthogranuloma. J Bone Miner Res 2017; 32: 784.
Wei YH, et al: Necrobiotic xanthogranuloma. Dermatol Ther 2015; 28: 7.
Wood AJ, et al: Necrobiotic xanthogranuloma. Arch Dermatol 2009; 145: 279.
Sarcoidosis
Sarcoidosis is a chronic multisystem inflammatory disease characterized by granuloma formation in most affected tissues. Sarcoidosis occurs worldwide, in patients of every age, ethnicity, and socioeconomic class, though prevalence and disease patterns vary. In Europe, it is most prevalent in Scandinavia, especially in Sweden, with a prevalence of 64 per 100,000 population. In the United Kingdom, the rate is 20 per 100,000, and in France and Germany, about 10 in 100,000, with lower rates in Spain and Japan of 1.4 in 100,000. In the United States the southeastern states and certain urban centers (New York City; Detroit; Washington, DC) show the highest prevalence, and there is a marked racial variation, with a rate of 10.9 per 100,000 for white persons and 35.5 per 100,000 for African Americans. Women are affected slightly more often than men, with the highest incidence in African American women between ages 30 and 39. The lifetime risk for the development of sarcoidosis is 0.85% for white and 2.4% for black U.S. residents. The disease begins most frequently between ages 20 and 40, with a second peak at ages 65–69. Patients with late-onset sarcoidosis are five times more frequently women than men, have uveitis, and have specific skin lesions in one third of cases.
Interleukin-2 (IL-2)– and IFN-γ–secreting CD4+ helper T (Th) cells are important in causing lesions, as are other Th1 and Th17 cytokines. Several genetic associations have been made with sarcoidosis, but the underlying cause still remains a mystery. HLA-DRB1 variants are associated with an increased risk. HLA-DQB1*0201 and HLA-DRB1*0301 are strongly associated with acute disease and a good prognosis. HLA-B8/DR3 may be associated with Lofgren syndrome. Mutations in the promoter region of TNF are associated with erythema nodosum (EN) in sarcoidosis in Caucasians, and a variant in intron 1 of the lymphotoxin alpha (LTA) gene is associated with EN in female Caucasian sarcoidosis patients. Polymorphisms in the IL-23 receptor are associated with sarcoidal uveitis. ANXA11, BTNL2, and CCR5 gene polymorphisms have also been identified in specific populations as conferring risk for sarcoidosis.
Cutaneous involvement is present in 25%–30% of patients with sarcoidosis and may be classified as specific, which reveals granulomas on biopsy, or nonspecific, which represents reactive inflammation such as EN which when biopsied does not show classic sarcoidal granulomas. In about 20% of patients, the skin lesions appear before the systemic disease; in 50%, the skin and systemic lesions appear simultaneously; and in 30%, the skin lesions appear after the systemic disease, sometimes by as much as 10 years. This is often coincidental with the tapering of systemic corticosteroids for pulmonary sarcoidosis. The cutaneous manifestations of sarcoidosis are varied, and numerous morphologic lesion types have been described, including common morphologies (papules, nodules, plaques, subcutaneous nodules, tattoo- or scar-associated lesions, hypopigmented lesions) and less common morphologies (ichthyosiform, erythrodermic, ulcerative). Involvement of the face, particularly around the nose, eyes, and mouth, is common. Lesions can develop around prior scars, tattoos, or remote foreign body deposits (such as on the knees). Lupus pernio describes plaque or nodular sarcoidosis of the nose and central face which can develop scale, deeply infiltrate, damage underlying structures, and be disfiguring. Lesions of special sites such as genital, mucosal, alopecic, and nail disease may also uncommonly occur. Sarcoidal lesions are usually multiple, firm, and elastic when palpated. They extend to involve the entire thickness of the dermis. The overlying epidermis may be slightly thinned, discolored, telangiectatic, or scaly. The color may be pink, red, or violaceous in some patients, but is frequently faint, showing dull tints of red, purple, brown, or yellow, according to the stage of development, and perhaps varying by the underlying skin tone. Usually, the lesions are asymptomatic, but approximately 10%–15% of patients itch. There is a racial difference in the frequency of cutaneous lesions in sarcoidosis. Among white patients, EN is as common as the specific cutaneous manifestations, and both types of cutaneous involvement occur in about 10% of white patients with sarcoidosis. In black patients, EN is much less common; however, specific cutaneous manifestations occur in 50% or more of patients. The skin lesions in general do not correlate with the extent or nature of systemic involvement or with prognosis. The exceptions are EN, which is associated with a good prognosis, and subcutaneous sarcoidosis and lupus pernio. The morphologic types of sarcoidosis are discussed next, and when possible, the relationship to systemic sarcoidosis.
Erythema Nodosum in Sarcoidosis
Erythema nodosum is the most common nonspecific cutaneous finding in sarcoidosis. EN rarely occurs in sarcoidosis beginning after age 65. Sarcoidosis may first appear with fever, polyarthralgias, uveitis, bilateral hilar adenopathy, fatigue, and EN. This combination, known as Lofgren syndrome, occurs frequently in Scandinavian whites and is rare in American blacks. The typical red, warm, and tender subcutaneous nodules of the anterior shins are distinctive and are most frequently seen in young women. The face, upper back, and extensor surfaces of the upper extremities may less frequently be involved. There is a strikingly elevated ESR, frequently above 50 mm/hr. EN is associated with a good prognosis, with the sarcoidosis involuting within 2 years of onset in 80% of patients. Conversely, the absence of EN is a risk factor for persistent disease activity. Sweet syndrome may also rarely be seen in association with sarcoidosis as a nonspecific finding.
Papular Sarcoid
Papules are the most common morphology of cutaneous sarcoidosis and are usually less than 1 cm in diameter. Lesions may be localized or generalized, in which case small papules predominate ( Fig. 31.9 ). This is also known as miliary sarcoid. The papules are especially numerous over the face, eyelids, neck, and shoulders. Plaques may occur by the expansion or coalescence of papules. In time, the lesions involute to faint macules. Hyperkeratosis may rarely be prominent, giving the lesions a verrucous appearance. “Papular sarcoidosis of the knee” is distinctive, in that disease may be limited to this site. In this region, the sarcoidal granulomas often contain foreign bodies, potentially representing remote childhood injury. In Caucasians, it often occurs in the context of Lofgren syndrome (see earlier) and has a good prognosis. Papular lesions along the alar rim in African Americans, in contrast, may be the first evidence of lupus pernio (see later) and portend a poor prognosis.
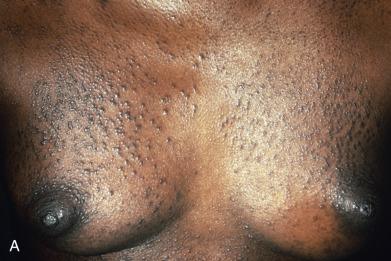
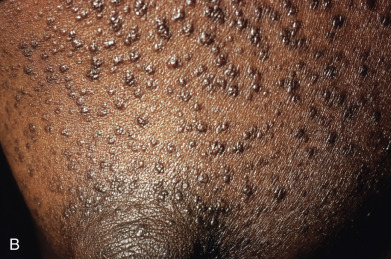
Annular Sarcoidosis
Papular lesions may coalesce or be arranged in annular patterns, usually with a red-brown hue ( Fig. 31.10 ). On palpation, the lesions are indurated. Central clearing with hypopigmentation, atrophy, and scarring may occur. Lesions favor the head and neck and are usually associated with chronic sarcoidosis. Alopecia may result in the center of the lesion. Annular plaques of sarcoidosis can preferentially develop in sun-exposed areas. This should be distinguished from annular elastolytic granulomas (see earlier).
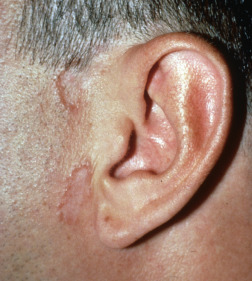
Hypopigmented Sarcoidosis
Hypopigmentation may be the earliest sign of sarcoidosis and is usually diagnosed in darkly pigmented races. Lesions vary from a few millimeters to more than 1 cm in diameter and favor the extremities. Although they appear macular, a dermal or subcutaneous component is often palpable.
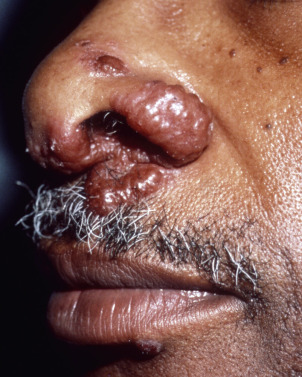
Lupus Pernio
Lesions typically are brown to violaceous, smooth, shiny plaques on the head and neck, especially the nose ( Fig. 31.11 ), cheeks, lips, forehead, and ears. They can be very disfiguring. Involvement of the nasal mucosa and underlying bone may occur and lead to nasal perforation and collapse of the nasal bridge resembling granulomatosis with polyangiitis. Upper aerodigestive tract involvement is also common. Ear, nose, and throat (ENT) evaluation is recommended. In three quarters of lupus pernio patients, chronic fibrotic respiratory tract involvement is found. In 43%, lupus pernio is associated with granulomas in the bones (punched-out cysts), most often of the fingers. Chronic ocular lesions occur in 37% of patients. Sarcoid involving the sinus is associated with lupus pernio in 50% of cases. Lupus pernio is typically seen in women in their fourth or fifth decade. The skin lesions rarely involute spontaneously. At times, lupus pernio may resemble rhinophyma. It is important to make the correct diagnosis, because ulceration of sarcoidal lesions may occur with laser treatment, even with pulsed dye laser. Patients with lupus pernio tend to have a chronic, recalcitrant course, and may require specific treatment based on this morphology (see later).
Ulcerative Sarcoidosis
Ulcerative sarcoidosis is very rare, affecting about 0.5% of patients with sarcoidosis. It affects primarily blacks, but it is also well recognized in Japanese. It is two to three times more common in women than men. In one third of cases, it is the presenting finding of sarcoidosis, except in Japan, where it is usually a late finding in patients with known sarcoidosis. The ulcerations may occur de novo or in sarcoidal plaques. Lesions favor the lower extremities, but most patients have lesions in more than one anatomic region. Trauma may be the inciting event. The clinical appearance may not be specific, but skin biopsies are diagnostic. Lupus pernio may also be present. Many patients have multisystem sarcoidosis, although infrequently, no other evidence of sarcoidosis is found. Biopsies may show necrosis in the center of sarcoidal granulomas. NL and ulcerative sarcoidosis share overlapping clinical features and the histology may overlap in some cases. Occasionally differentiating the two requires evaluation for extracutaneous disease, which is present in sarcoidosis and absent in NL. Methotrexate, which can be therapeutic in sarcoidosis, may also lead to ulceration in sarcoidosis patients.
Subcutaneous Sarcoidosis
Subcutaneous sarcoidosis is also known as Darier-Roussy sarcoid and consists of a few to numerous 0.5–3 cm, deep-seated nodules on the trunk and extremities; only rarely do they appear on the face. The overlying epidermis may be normal (30%), erythematous (50%), or slightly violaceous (10%). The lesions are usually asymptomatic. About 90% of patients will have multiple lesions, and the upper extremity is most frequently affected (virtually 100% of patients). Lesions on the upper extremity have a tendency to form indurated linear bands from the elbow to the hand on the cubital side of the forearm. The amount of subcutaneous involvement in the upper extremity may be so extensive as to simulate chronic cellulitis. A biopsy is usually required to confirm the diagnosis. Although it has been described that 90% of patients with subcutaneous sarcoid also will have systemic involvement, usually bilateral hilar adenopathy, it should be noted that sarcoidosis is by definition a multiorgan disease, and hilar adenopathy is the most common lung finding—it remains unclear whether specific cutaneous morphologies such as subcutaneous lesions confer prognosis, beyond that noted for lupus pernio.
Plaques
These distinctive lesions are flat-surfaced, slightly elevated plaques ( Fig. 31.12 ) that appear with greatest frequency on the cheeks, limbs, and trunk symmetrically. Superficial nodules may be superimposed, and coalescence of plaques may lead to serpiginous lesions. Involvement of the scalp may lead to permanent alopecia. The finding of alopecia in an annular plaque with a raised border should raise the diagnostic consideration of sarcoidosis.

Erythrodermic Sarcoidosis
Erythrodermic sarcoidosis is an extremely rare form of sarcoidosis. A diffuse infiltrative erythroderma of the skin usually begins as erythematous, scaling patches that merge to involve large portions of the body. A biopsy is confirmatory, but the diagnosis can be clinically suspected if small, “apple jelly” papules are seen on diascopy throughout the erythroderma. Diffuse granulomatous dermatitis in the setting of myelodysplastic disorders should be excluded.
Ichthyosiform Sarcoidosis
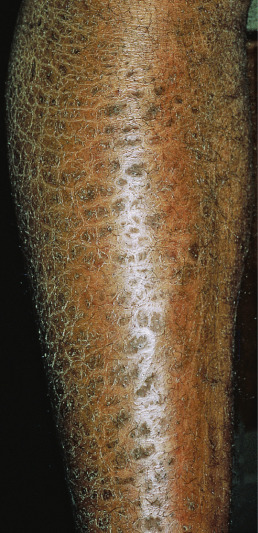
Ichthyosiform sarcoidosis resembles ichthyosis vulgaris or acquired ichthyosis, with fine scaling usually on the distal extremities ( Fig. 31.13 ). It is virtually always seen in nonwhite persons, especially African Americans. Almost all patients have or will develop systemic disease. In 75% of patients, the skin lesions follow or occur at the same time as the diagnosis of systemic sarcoidosis. Although the lesions have no palpable component, a biopsy will reveal dermal noncaseating granulomas.