Lichen Planus
Lichen planus (LP) is a common, pruritic, inflammatory disease of the skin, mucous membranes, and hair follicles. It occurs throughout the world, in all races. Cutaneous LP affects 0.3% of men and 0.1% of women. Oral LP affects 1.5% of men and 2.3% of women. It may be familial in rare cases. The pattern of LP detected and the age distribution vary among various genetic and geographic groups; certain human leukocyte antigen (HLA) alleles (HLA-DR/DQ, HLA-A3), gene mutations (MTHFR), and single-nucleotide polymorphisms (tumor necrosis factor–α [TNF-α], NRP2, IGFBP4) have been reported to confer risk in select populations and subgroups. In persons of European descent, LP appears primarily after age 20 and peaks between 40 and 70. Very few cases appear after age 80. Childhood LP typically accounts for 5% or less of LP cases, although in some regions, including the Indian subcontinent, Arab countries, and Mexico, it represents 10%–20%. Race appears to be the critical factor; in the United States, LP occurs more commonly in African American children, and in the United Kingdom, for example, Indians account for 80% of childhood LP. Pathophysiologically LP is a T-cell disorder with increased Th1 cytokine expression and T-cell activity at the basement membrane zone.
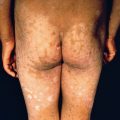
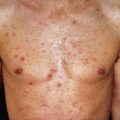
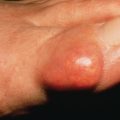
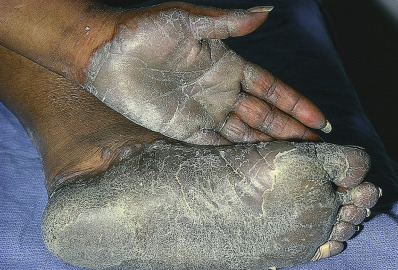
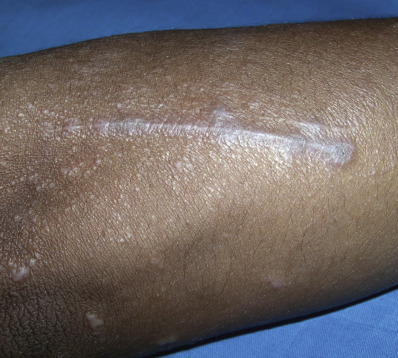
The primary lesions of LP are characteristic, almost pathognomonic: small, flat-topped, polygonal papules ( Fig. 12.1 ). The color of the lesions initially is erythematous. Well-developed lesions are violaceous, and resolving lesions are often hyperpigmented, especially in patients with darker skin. The surface is glistening and dry, with scant, adherent scales. On the surface, gray or white puncta or streaks (Wickham striae) cross the lesions—a feature seen more easily with dermoscopy. Lesions begin as pinpoint papules and expand to 0.5–1.5 cm plaques. Infrequently, larger lesions are seen. There is a predilection for the flexor wrists, trunk, medial thighs, shins, dorsal hands, and glans penis ( Fig. 12.2 ). The face is only rarely involved, with lesions usually confined to the eyelids and lips. The palms and soles may be affected with small papules or hyperkeratotic plaques ( Fig. 12.3 ). Certain morphologic patterns favor certain locations (e.g., annular lesions favoring penis; keratotic lesions favoring anterior shins). Lesions are often bilateral and relatively symmetric. The Koebner phenomenon occurs in LP ( Fig. 12.4 ). Oral mucosal involvement is common.
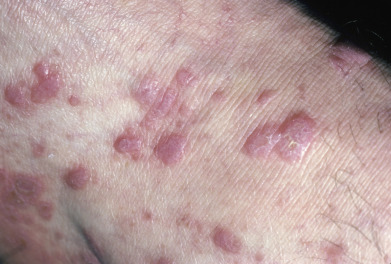
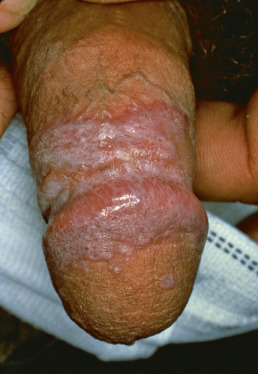
Pruritus is often prominent in LP. The pruritus may precede the appearance of the skin lesions, and the intensity of the itch may seem out of proportion to the amount of skin disease. Most patients react to the itching of LP by rubbing rather than scratching, and thus scratch marks are usually not present, though linear papules of LP can indicate prior scratching and koebnerization.
The natural history of LP is highly variable and dependent on the site of involvement and the clinical pattern. Two thirds of patients with skin lesions will have LP for less than 1 year, and many patients spontaneously clear in the second year. Hypertrophic and mucosal disease tends to be more chronic. Recurrences are common. In older patients, and those with comorbidities such as hyperlipidemia and diabetes, LP may persist for longer duration.
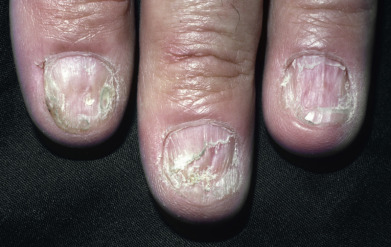
Nail changes are present in approximately 5%–10% of patients ( Fig. 12.5 ), with fingernails more often affected than toenails. Involvement of the nail can occur as an initial manifestation, especially in children. Longitudinal ridging and splitting are most common, seen in 90% of patients with nail involvement. Onycholysis and subungual debris may be present, indicating involvement of the nail bed. The lunulae are red in 30% of patients with nail LP. Involvement of the entire matrix may lead to obliteration of the whole nail plate (anonychia). Pterygium formation is characteristic of LP of the nails, but seen in only about 20% of patients. The nail matrix is destroyed by the inflammation and replaced by fibrosis. The proximal nailfold fuses with the proximal portion of the nail bed. Dermoscopy can show pitting of the nail matrix, tachyonychia, chromonychia, onycholysis, or splinter hemorrhages. LP may be a cause of some cases of 20-nail dystrophy of childhood. In the absence of periungual lesions or pterygium formation, 20-nail dystrophy usually resolves spontaneously, and frequently in these children, no other stigmata of cutaneous or mucosal LP are found.
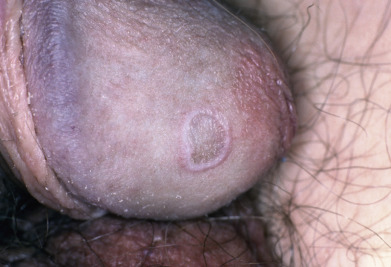
Involvement of the genitalia, with or without lesions at other sites, is common. On the glans or shaft of the penis, the lesions may consist of flat, polygonal papules, or these may be annular. Erosive LP can occur on the glans. Simultaneous involvement of the gingival and penile mucosa may occur. On the labia and anus, similar lesions are observed, which may be whitish because of maceration. Half of women with oral LP also have vulvovaginal LP, but in only half of these patients is the genital LP symptomatic. Vulval LP occurs in three main forms. The classic type presents with polygonal papules resembling cutaneous LP and affects the clitoral hood and labia minora. Pruritus is the usual symptom. Although only about 20% of women with vulval LP have erosive or ulcerative LP, this type represents the vast majority of patients seen for vulval LP, because it is usually very symptomatic. Soreness, pain, and dyspareunia are frequent complaints. Vaginal involvement with a bloody discharge can occur. Involvement is symmetric from the fourchette to the anterior vestibule. The erosions have a lacy white periphery, a good area to biopsy to confirm the diagnosis of vulval LP. Vulvovaginal-gingival syndrome is characterized by involvement at these three sites, with significant long-term sequelae due to scaring; patients may also have scarring at other mucosal sites, including the esophagus and eye. The third, and least common, form of vulval LP is the hypertrophic type. It involves the perineum and perianal skin (but not the vagina) with warty plaques with a violaceous edge. Pruritus is severe. Vulval splitting, vaginal stenosis, and sealing of the clitoral hood may be caused by LP. It is important to distinguish vulvovaginal LP from lichen sclerosus et atrophicus.
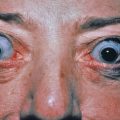
Conjunctival involvement is a rarely recognized complication of LP but was seen in 0.5% of patients with vulval LP in one series. It most frequently occurs in patients with involvement of other mucosal surfaces. Cicatrization, lacrimal canalicular duct scarring, and keratitis can occur. It may closely simulate mucous membrane pemphigoid. Routine histology and direct immunofluorescence (DIF) may be required to confirm the diagnosis.
Otic involvement by LP is rarely reported. It affects primarily females (80% of patients) and is associated with oral and vulval LP in more than 50% of cases, and cutaneous LP in 5 of 19 (26%). Hearing loss and external auditory canal stenosis are the most common otic complaints and complications. Four of 19 (21%) patients with otic LP also had esophageal involvement.
LP of the esophagus is uncommon but increasingly being recognized, and the diagnosis is frequently delayed. One study involving endoscopy in 32 consecutive patients with LP identified 10 with probable and 10 with definitive esophageal disease. Dysphagia, odynophagia, and weight loss are typical manifestations. The proximal esophagus is more frequently affected. Most patients have coexistent oral disease. Esophageal involvement is much more common in women. One paper in the gastrointestinal literature noted that stricture formation occurs in 80% of esophageal LP and may require frequent dilations; the observed rate in dermatologic practice is much lower. Esophageal squamous cell carcinoma (SCC) may complicate esophageal LP, suggesting that, once this diagnosis is made, routine gastrointestinal evaluation is required.
Whether the many clinical variants of LP represent separate diseases or part of the LP spectrum is unknown. They all demonstrate typical LP histologically. The variants are described separately because their clinical features are distinct from classic LP. Some patients with these clinical variants may have typical cutaneous, follicular, or mucosal LP. The more common or well-known variants are described here.
Linear Lichen Planus
Small, linear lesions caused by the Koebner phenomenon often occur in classic LP. Limitation of LP to one band or streak has also been described in fewer than 1% of patients of European descent. In Japan, however, up to 10% of cases are linear, and in India, 7% of childhood cases of LP are linear. Although originally described as following dermatomes (zosteriform), the lesions actually follow lines of Blaschko. It is more common in children but also occurs in adults. Papules with varying degrees of overlying hyperkeratosis or simple hyperpigmentation may be the presenting manifestations. There are often “skip areas” of normal skin between the individual lesions. Ipsilateral mucosal disease may be seen.
Annular and Annular Atrophic Lichen Planus
Annular LP occurs in 3%–7% of patients with LP, and is more common in men (90% of cases). Lesions with this configuration favor the axilla, penis/scrotum ( Fig. 12.6 ), and groin. LP lesions of the mucosa, scalp, and nails are rare in patients with annular LP. Patients usually have fewer than 10 lesions. Most patients with annular LP are asymptomatic. The ringed lesions are composed of small papules and measure about 1 cm in diameter. Central hyperpigmentation may be the dominant feature. They may coalesce to form polycyclic figures. Annular lesions may also result from central involution of flat papules or plaques, forming lesions with violaceous, elevated borders and central hyperpigmented macules. A rare type of annular lichenoid dermatitis which may resemble morphea, inflammatory vitiligo, or mycosis fungoides has been described in pediatric patients termed annular lichenoid dermatitis of youth; one report from a region with endemic borrelia suggested a possible etiologic link, which has not been proven.
Hypertrophic Lichen Planus
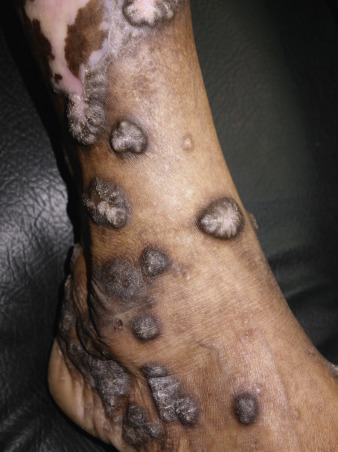
Hypertrophic LP usually occurs on the shins but may occur anywhere. The anterior lower leg below the knee is the sole area of involvement in most patients. The typical lesions are verrucous plaques with variable amounts of scale ( Fig. 12.7 ). At the edges of the plaques, small, flat-topped, polygonal papules may at times be discovered. Superficial inspection of the lesion often suggests psoriasis or a keratinocytic neoplasm rather than LP, but the typical appearance resembling rapidly cooled igneous rock (igneous rock sign) may be useful in suggesting LP over keratinocytic neoplasms. The lesions are of variable size but are frequently several centimeters in diameter and larger than the lesions of classic LP. Dermoscopy of hypertrophic LP may demonstrate pearly white areas and peripheral striations, which can help distinguish it from mimickers in some cases; however, clinical diagnosis may be difficult, and biopsy is often required. Histologically, the pseudoepitheliomatous keratinocyte hyperplasia may be marked, leading to the erroneous diagnosis of SCC. Eosinophils are much more often present in the dermal infiltrate of hypertrophic LP than classic LP. True SCC may also evolve from long-standing hypertrophic LP, over long as 11–12 years. In addition, keratoacanthoma-like proliferations may occur in lesions of hypertrophic LP. This has also been called “hypertrophic lichen planus–like reactions combined with infundibulocystic hyperplasia.” Hypertrophic LP is chronic and often refractory to topical therapy. Hypertrophic lupus erythematosus (LE) resembles hypertrophic LP both clinically and histologically. Hypertrophic LE tends to affect the distal extremities, face, and scalp. The finding of continuous granular immunoglobulin on DIF strongly suggests a diagnosis of hypertrophic LE rather than LP.
Erosive/Ulcerative/Mucosal Lichen Planus
Erosive LP has significant impact on quality of life, and patients with erosive LP have high levels of depression, anxiety, and stress. Ulcerative LP is rare on the skin but common on the mucous membranes. A rare ulcerative variant of cutaneous LP, or LE/LP overlap syndrome, affects the feet and toes, causing bullae, ulcerations, and permanent loss of the toenails. These chronic ulcerations on the feet are painful and disabling. Cicatricial alopecia may be present on the scalp, and the buccal mucosa may also be affected. These cases are a therapeutic challenge, and aggressive oral retinoid or immunomodulatory treatment is indicated if there is a poor response to standard topical and systemic agents. Skin grafting of the soles has produced successful results.
Oral mucosal LP is the most common form of mucosal LP, and it is usually chronic. Between 10% and 15% of patients with oral LP will also have skin lesions. Women represent 50%–75% of patients with oral LP. Oral LP in women begins 10 years later than in men (age 57 vs. 47). Oral lesions may be reticulate (reticular), the oral version of Wickham striae ( Fig. 12.8 ), erythematous (atrophic), or ulcerative (erosive) ( Fig. 12.9 ); lesions are often bilateral and symmetric. The most common pattern in oral LP is the ulcerative form (40% of patients). Usually, reticulate and erythematous lesions are found adjacent to the ulcerative areas. The erythematous pattern is the predominant pattern in 37% of patients, but almost always, reticulate lesions are also seen in these patients. In oral LP, the “classic” reticulate lesions are most prominent in 23% of patients. Rarely patients may have a white plaque mimicking leukoplakia, bullous, or papular lesions. Symptoms are least common in patients with reticulate lesions; 23% are symptomatic, and then only when the tongue is involved. All patients with erosive lesions are symptomatic, usually with burning or pain. Patients may simultaneously have several patterns, so patients are characterized by the primary form they exhibit. Lesions appear on any portion of the mouth, and multisite involvement is common. The buccal mucosa is involved in 90%, the gingiva in more than 50%, and the tongue in about 40%.
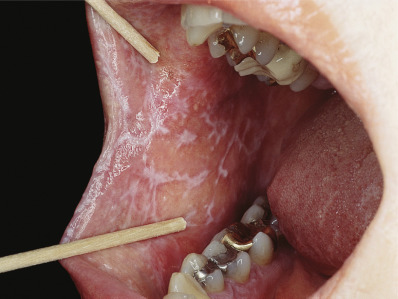
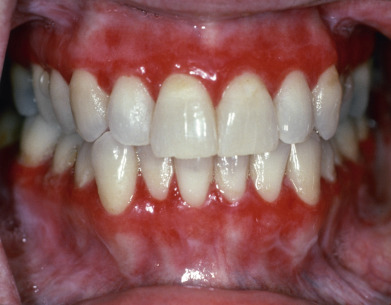
On the gingiva, LP may produce desquamative gingivitis (see Fig. 12.9 ). Gingival involvement is particularly difficult to diagnose and often requires biopsy for both histology and DIF to confirm the diagnosis and exclude other autoimmune causes of desquamative gingivitis. Gingival involvement is associated with accelerated gingival recession. Mechanical injury from dental procedures and poorly fitting appliances may trigger or exacerbate gingival LP. On the tongue and palate, lesions are often mistaken for leukoplakia. The lower lip is involved in 15% of oral LP patients, but the upper lip in only 2%. Lower lip LP is frequently mistaken for actinic cheilitis. Imiquimod treatment can lead to exacerbation of the labial LP, with extensive erosion and crusting. Oral LP is stable but chronic, with fewer than 3% of patients having a spontaneous remission in an average 5-year follow-up. Periodontitis appears to exacerbate oral LP, especially gingival disease. Plaque control either by the patient after training or by a dental professional improves the clinical appearance and pain.
Oral lichenoid lesion (OLL), or oral lichenoid reaction (OLR), refers to an oral lesion histologically identical to oral LP (OLP) but from a different cause, such as graft-versus-host disease (GVHD), medications (antihypertensives, dapsone, nonsteroidal antiinflammatory drugs [NSAIDs], penicillamine, phenothiazines, antimalarials, oral hypoglycemics, and more), and local or systemic exposures. Gold, cobalt, indium, manganese, chrome, nickel, palladium, cinnamate, and spearmint sensitivity may induce OLLs. The most common causes, however, are the metals in dental amalgams, including mercury, copper, zinc, and tin. If the lesions in the oral mucosa are physically close to the amalgam, removal of the amalgam will lead to resolution of the OLL in 36%. In patients patch test positive to a metal in the amalgam, 44% and 47% of OLLs healed with removal of the amalgam in two studies. In patients with the OLL in strict contact with the amalgam, and there is a relevant positive patch test to a component of the amalgam, 80% to 90% or more of OLLs will heal. Patch testing, however, may not identify all patients whose OLLs improve with removal of the oral metal. Rarely, patients with metal sensitivity will also have skin and nail lesions that improve with removal of the oral metal. In one study, 6 of 10 patients with nail LP who were patch test positive to a metal in their amalgam improved with removal of the dental material or with oral disodium chromoglycate treatment.
Involvement of the vulva and vagina with LP, along with the gingiva, has been called the vulvovaginal-gingival (VVG) syndrome. Although all three of these mucous membranes may be involved, only one or two sites may be involved at any one time. The prevalence of erosive vulvar LP had been underappreciated simply because many women with oral LP did not volunteer their vulvovaginal complaints and were not asked about them. The vaginal lesions of VVG are erythematous, friable erosions that are very painful. Untreated scarring is severe and can lead to adhesions, vestibular bands, and even vaginal stenosis. In one third of patients, typical reticulate buccal LP is seen, and in up to 80% the oral mucosa is also involved. Cutaneous lesions occur in 20%–40% of VVG patients. The course of the VVG syndrome is protracted, and patients frequently have sequelae, including chronic pain, dyspareunia, and even scarring of the conjunctiva, urethra, and oral, laryngeal, pharyngeal, and esophageal mucosae. Nails are involved in about 15% of patients with VVG, compared with only 2% of patients with oral LP. The VVG syndrome is now considered to be a separate subgroup of mucosal LP that is particularly disabling, scarring, and refractory to therapy.
Bullous Lichen Planus
Two forms of LP may be accompanied by bullae. In bullous LP (BLP), individual lesions, usually on the lower extremities, will vesiculate centrally, with bullae confined to preexisting LP lesions. Histology reveals the bullae are due to a large Max-Joseph space within the extensive lichenoid inflammatory infiltrate. These lesions often spontaneously resolve. There are familiar forms of bullous lichen planus, with earlier onset and more chronic course. Recently checkpoint inhibitors such as PD-1 inhibitors (pembrolizumab) have been shown to cause bullous LP-like reactions, LP pemphigoides, and bullous pemphigoid.
LP pemphigoides describes a rare subset of patients who usually have typical LP, then, an average of 8 months later, develop blistering on their LP lesions and on normal skin. Less often, the blister and the LP lesions occur simultaneously. Clinically, these patients appear to be a combination of LP and bullous pemphigoid. Oral disease may occur and resemble either LP or mucous membrane pemphigoid. LP pemphigoides can be triggered by medications, especially angiotensin-converting enzyme (ACE) inhibitors, as well as interferon (IFN), hepatitis B virus (HBV), and psoralen plus ultraviolet A (PUVA). Pruritus may be severe, and lesions may evolve to resemble pemphigoid nodularis. Bullous pemphigoid affects an older age-group than LP pemphigoides; typical onset for LP pemphigoides is 30–50. Histologically, the LP lesions show LP, and the bullous lesions show the features of bullous pemphigoid. DIF is positive in a linear pattern, with immunoglobulin G (IgG) and C3 along the basement membrane zone (BMZ), at the roof of saline split skin. The antigen targeted by the autoantibody in LP pemphigoides is located in the same region as the bullous pemphigoid antigen, at the basal hemidesmosome. Antibodies from patients with LP pemphigoides typically bind the 180-kD bullous pemphigoid antigen, but in a different region from bullous pemphigoid sera. One case of clinical and histologic bullous LP was reported with autoantibodies to BP180, with authors suggesting chronic inflammation and damage to the BMZ may expose antigens and lead to development of overt LPP over time. LP pemphigoides tends to follow a benign and chronic course, even compared with bullous pemphigoid. Treatment of LP pemphigoides is similar to bullous pemphigoid, with potent topical steroids, systemic steroids, tetracycline, nicotinamide, intravenous immune globulin (IVIG), and immunosuppressives all being variably effective.
Hepatitis-Associated Lichen Planus
Three liver conditions have been associated with LP: hepatitis C virus (HCV), hepatitis B immunization, and primary biliary cirrhosis. HCV infection has been found in proportionately more patients with LP than in controls in numerous studies. The prevalence of HCV infection in patients with LP varies from 1.6% to 20%. There is an association with the HLA-DR6 allele. A systematic review and meta-analysis of 1807 cases of oral LP and 2519 controls revealed an odds ratio was 6.07, with OLP patients having a sixfold higher risk for HCV infection than controls. Despite multiple systematic reviews suggesting a connection, the association of HCV infection and LP has been questioned, perhaps due to geographic differences in disease prevalences. In a large series of patients with oral LP from the United States, none of the 195 patients was infected with HCV, whereas 29% of patients with oral LP from Italy had HCV. In Scotland, 20% of patients infected with HCV had oral LP, compared with 1% of seronegative patients. Although the data are conflicting, screening for HCV appears appropriate in persons from a geographic region where or a population in whom HCV infection is frequently associated with LP. The clinical features of LP in patients with hepatitis C are identical to classic LP, but LP patients with HCV infection are reported as being more likely to have erosive mucous membrane disease, and higher rates of malignancy. The HCV genome is not found in lesions of LP associated with HCV infection. The existence of underlying hepatitis cannot be predicted by clinical pattern or the results of liver function tests. Treatment of hepatitis C with IFN-α may be associated with the initial appearance of LP or exacerbation of preexisting LP. LP may occur at IFN injection sites, and skin testing may reproduce LP-like lesions. LP may improve or may not change with IFN and ribavirin treatment for hepatitis C. Improvement is usually seen toward the end of the treatment course. Most patients do not completely clear their LP. Treatment with newer antivirals for HCV, including ledipasvir-sofosbuvir, have shown conflicting results, with some responders clearing their coexistent LP, and one case of LP developing after HCV clearance.
Although less robust data support an association, HBV immunization may be associated with the appearance of LP in both children and adults. Lesions are typical of LP, and the oral mucosa may be affected. Typically, the first lesions of LP appear about 1 month after the second dose of vaccine. Lesions usually resolve after some time.
Primary biliary cirrhosis and LP may coexist. Patients with this liver abnormality also have a marked propensity to develop a lichenoid eruption while receiving d -penicillamine therapy. Xanthomas in patients with primary biliary cirrhosis may appear initially in lesions of LP, and the infiltrate, although lichenoid, may contain xanthomatous cells. Primary sclerosing cholangitis has been associated with oral LP.
Cancer Risk and Lichen Planus
The risk of malignant transformation in LP varies by morphologic type, with mucosal disease having higher rates. Rare cases of SCC of the skin occurring on the lower leg in lesions of hypertrophic LP have been reported. Cutaneous LP alone is not considered to be a condition with increased cancer risk. Oral LP and vulvovaginal LP, however, do appear to increase the risk of developing SCC. In a study of 13,100 women in Finland, LP was associated with an increased risk of cancers of the lip, tongue, oral cavity, esophagus, larynx, and vulva. About 1% of patients with oral LP will develop oral SCC. A higher risk is seen in smokers, alcoholics, and patients with HCV infection. SCC occurs in patients with erythematous or ulcerative LP, not in those with only the reticulate pattern. Of the oral LP patients who develop oral SCC, only about 45% have one cancer. The majority develop multiple cancers, and close vigilance is recommended in these patients. LP patients with erosive penile and vaginal disease also have developed SCC. The number of penile cases is too low to determine the frequency, but in patients with vulvar LP, development of SCC may be as high as 3%. Clinicians should have a low threshold to biopsy fixed erosive or leukokeratotic lesions in patients with mucosal LP. The use of oral and topical calcineurin inhibitors (CNIs) for LP has been associated with the appearance of SCC on the genitalia. There is no evidence that the medications caused the neoplasia, but if these agents are used, regular follow-up and careful examination are required.
Pathogenesis and Histology
LP is characterized by a Th1 immunologic reaction mediated by CD8+ T cells. These cells induce keratinocytes to undergo apoptosis. Although this inflammatory reaction is thought to be autoimmune, the antigen targeted by these effector T lymphocytes is unknown. The prevalence of autoimmune phenomena is not increased in patients with classic cutaneous LP, however, patient with erosive LP of the vulva (and lichen sclerosus) may have an autoimmune basis. A personal and family history of autoimmune disorders (usually thyroid disease) is present in up to 30% of patients with vulvar LP, and up to 40% have circulating autoantibodies. Patients with oral LP may also have elevated rates of thyroid disease. Patients with LP and OLP have a high rate of dyslipidemia, with elevated triglycerides, elevated low-density lipoprotein (LDL) cholesterol and reduced high-density lipoprotein (HDL) cholesterol. Inflammatory markers also are elevated in the blood of LP patients, potentially contributing to this dyslipidemia. In addition, both insulin resistance and frank type 2 diabetes mellitus are increased in patients with LP compared with controls. Both insulin resistance and dyslipidemia are cardiovascular risk factors. Adult LP patients should be evaluated appropriately. Drug induced lichenoid eruptions are common with a long list of potential triggering agents (including but not limited to checkpoint inhibitors, NSAIDs, allopurinol, oral hypoglycemic agents, anticonvulsants, cardiovascular/blood pressure medications, biologics, antimicrobials, antimalarials, antipsychotic drugs, and more, with some causing specifically photo-accentuated lichenoid eruptions, including antiviral agents such as lepidasvir/sofosbuvir).
LP pemphigoides is hypothesized to result from exposure to the immune system of epitopes in the BP180 antigen as keratinocytes are destroyed by the lichenoid inflammation. Epitope spreading can occur, and LP pemphigoides patients may also develop autoantibodies to the same epitopes as bullous pemphigoid patients.
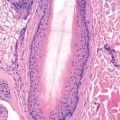
The histologic features of LP are distinctive and vary with the stage of the lesion. In early lesions, there is an interface dermatitis along the dermoepidermal junction. As the lesion evolves, the epidermis takes on a characteristic appearance. There is destruction of the basal layer with a “sawtooth” pattern of epidermal hyperplasia, orthokeratosis, and beaded hypergranulosis. The basal cells are lost, so the basal layer is described as “squamatized.” In the superficial dermis, there is a dense, bandlike infiltrate composed of lymphocytes and melanophages. “Civatte bodies” (cytoid bodies, colloid bodies) represent necrotic keratinocytes in the superficial dermis. Hypertrophic LP shows marked epidermal hyperplasia (pseudoepitheliomatous hyperplasia). Old lesions of LP show effacement of the rete ridge pattern, melanophages in the upper dermis, and occasional Civatte bodies. LP rarely demonstrates parakeratosis or eosinophils (except in hypertrophic LP, where these are characteristic). The presence of either of these suggests a different cause of lichenoid tissue reaction, such as lichenoid drug eruption. Lichen planopilaris, frontal fibrosing alopecia (FFA), and Graham-Little-Piccardi-Lasseur syndrome show the findings of LP, centered on the superficial follicular epithelium.
Lesions of LP of the skin or mucosae can demonstrate clumps of IgM on DIF, and less frequently IgA, IgG, and C3, subepidermally, corresponding to the colloid bodies. Dense, shaggy staining for fibrinogen along the BMZ is characteristic of LP. A lichenoid drug eruption may be difficult to differentiate from LP. The presence of eosinophils or parakeratosis supports the diagnosis of lichenoid drug eruption. GVHD tends to have a sparser infiltrate than LP, and the infiltrate may involve hair follicles if present. Hypertrophic LE may be histologically identical to LP, and the diagnosis is best made by clinical correlation, serologies, and DIF. In most other forms of LE, there is a greater tendency for epidermal atrophy with parakeratosis, dermal mucin is found, and follicular plugging is more prominent. The infiltrate in lupus tends to surround and involve deep portions of the appendageal structures, such as the follicular isthmus and eccrine coil. Deep, nodular, perivascular lymphoplasmacytic infiltrates and necrosis of the fat lobule with fibrin or hyalin rings are also findings characteristic of LE.
Differential Diagnosis
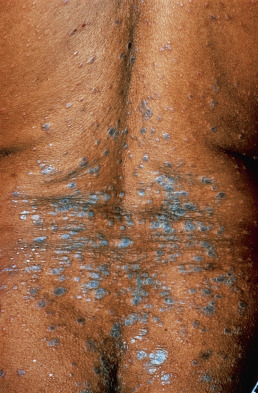
Classic LP ( Fig. 12.10 ) displays lesions that are so characteristic that clinical examination is often adequate to lead to suspicion of the diagnosis. Lichenoid drug eruptions may be difficult to distinguish. A lichenoid drug reaction should be suspected if the eruption is photodistributed, scaly but not hypertrophic, and confluent or widespread—clinical features that are unusual for idiopathic LP. The presence of oral mucosa involvement may prompt suspicion of LP, but oral lesions may occasionally occur in lichenoid drug eruptions as well. Pityriasis rosea, guttate psoriasis, the small papular or lichenoid syphilid, and pityriasis lichenoides et varioliformis acuta are dermatoses that may resemble generalized LP. Mucous membrane lesions may be confused with leukoplakia, LE, mucous patches of syphilis, candidiasis, cancer, and oral lesions of autoimmune bullous diseases, such as pemphigus or cicatricial pemphigoid. On the scalp, the atrophic lesions may be mistaken for other cicatricial alopecias, such as LE, folliculitis decalvans, and pseudopelade of Brocq. Hypertrophic LP type may simulate psoriasis, LE, SCC, and keratoacanthomas. Isolated patches of LP may resemble lichen simplex chronicus.
Treatment
There is virtually no high-quality evidence for treatment of LP of the skin, scalp, or mucosae. Limited lesions may be treated with superpotent topical corticosteroids or intralesional steroid injections. Topical tacrolimus or pimecrolimus can be effective, particularly for erosive disease. Topical vitamin D derivatives have also been reported to help in small studies. In patients with widespread disease, these treatments are usually unsatisfactory. Widespread lesions respond well to systemic corticosteroids but tend to relapse as the dose is reduced. Monthly pulse dosing has been championed by dermatologists in India. Phototherapy may be effective for cutaneous LP, including narrow-band (NB) ultraviolet B (UVB), UVA I, and PUVA (topical or oral). NB UVB was superior to systemic corticosteroids in one study. A variety of lasers have been reported as helpful in isolated case reports for different morphologic and anatomic forms of LP. Photodynamic therapy with topical 5-aminolevulinic acid can be effective in genital LP. The oral retinoids—isotretinoin, alitretinoin, and acitretin, in doses similar to or slightly lower than those used for other skin conditions—may also be useful and avoid the long-term complications of systemic steroids. They are especially beneficial in patients with hypertrophic LP and palmoplantar LP. Retinoid therapy may be combined with phototherapy in refractory cases. Hydroxychloroquine in standard doses can be effective for cutaneous, oral, genital and follicular LP. Adding quinacrine, 100 mg daily, may be considered in patients with only a partial response to hydroxychloroquine. Thalidomide, 50–150 mg daily, can improve refractory oral and cutaneous LP. Griseofulvin can improve cutaneous LP. Sulfasalazine led to a partial response in 11 of 26 patients. Low-molecular-weight heparin (enoxaparin), injected subcutaneously once a week, led to remission of cutaneous and reticulate oral LP in 61% of patients and improvement in 11%. Enoxaparin is less effective than systemic steroids. Dapsone and metronidazole in small studies failed to outperform topical or phototherapy, though one small series did show 40% of patients responding to metronidazole 250 mg every 8 hours. Apremilast, a phosphodiesterase type IV inhibitor, at a dose of 20 mg twice daily, showed modest efficacy in pooled data, but pruritus was dramatically decreased.
In the most severe cases, immunosuppressive agents may be indicated. Cyclosporine, methotrexate, and mycophenolate mofetil are all options and can induce remission in severe cases of cutaneous and oral LP. The TNF inhibitors have been effective in anecdotal cases. Similarly, extracorporeal photophoresis (ECP), anakinra, rituximab, systemic tacrolimus, and IVIG have been successful in extremely refractory cases.
For oral lesions, superpotent steroids in Orabase or gel form are useful. Vinyl dental trays may be used to apply steroid ointments to the gingiva. Begin with 30-minute applications three times a day and reduce to maintenance of 20 minutes every evening. Addition of nystatin to clobetasol in Orabase may be especially effective. Overall, more than 70% of patients with vulvar LP have relief of symptoms with topical clobetasol. Intralesional injections may be used for focal unresponsive lesions. Topical tacrolimus 0.1% ointment has become standard treatment in erosive LP of the oral and genital mucosa. Burning may occur initially but can be reduced by concomitant use of topical steroids or initial use of a lower strength of tacrolimus ointment. Higher concentrations, up to 0.3%, also may be used. Most patients have a partial but significant response, with increased ability to eat with much less pain. Blood levels can be detected, independent of area of involvement, but tend to decrease over time as the oral erosions heal. Pimecrolimus can be used successfully in patients intolerant of topical tacrolimus. Sustained remissions are rare, and chronic use is usually required to maintain remission. Topical cyclosporine is comparable to topical steroids and should not be used first-line, but may be tried in recalcitrant cases. Topical retinoids may be used but are generally less desirable than topical steroids. Aloe vera gel has been used and in one study was equivalent to steroids. PUVA, photodynamic therapy, and 308-nm excimer laser have been effective in oral LP. The systemic agents recommended earlier to treat cutaneous LP may also improve mucosal disease. For VVG syndrome, corticosteroids topically and systemically are beneficial. Topical therapy with corticosteroids may be enhanced by mixing the steroid in vaginal bioadhesive moisturizer (Replens). Iontophoresis may improve delivery.
Aghbari SMH, et al: Malignant transformation of oral lichen planus and oral lichenoid lesions. Oral Oncol 2017; 68: 92.
Ahlgren C, et al: Contact allergy to gold in patients with oral lichen lesions. Acta Derm Venereol 2012; 92: 138.
Alaizari NA, et al: Hepatitis C virus infections in oral lichen planus. Aust Dent J 2016; 61: 282.
Alomari A, McNiff JM: The significance of eosinophils in hypertrophic lichen planus. J Cutan Pathol 2014; 41: 347.
Alrashdan MS, et al: Oral lichen planus. Arch Dermatol Res 2016; 308: 539.
Atzmony L, et al: Treatments for cutaneous lichen planus. Am J Clin Dermatol 2016; 17: 11.
Brauns B, et al: Intralesional steroid injection alleviates nail lichen planus. Int J Dermatol 2011; 50: 626.
Cesinaro AM: Annular lichenoid dermatitis (of youth). Am J Dermatopathol 2017; ePub ahead of print.
Cohen PR, Kurzrock R: Anakinra-responsive lichen planus in a woman with Erdheim-Chester disease. Dermatol Online J 2014; 20: 21241.
Domingues E, et al: Imiquimod reactivation of lichen planus. Cutis 2012; 89: 276.
Dreijer J, et al: Lichen planus and dyslipidaemia. Br J Dermatol 2009; 11: 626.
Friedman P, et al: Dermoscopic findings in different clinical variants of lichen planus. Dermatol Pract Concept 2015; 5: 51.
Fox LP, et al: Lichen planus of the esophagus. J Am Acad Dermatol 2011; 65: 175.
Fujii M, et al: Bullous lichen planus accompanied by elevation of serum anti-BP180 autoantibody. J Dermatol 2017; 44: e124.
Garcia-Pola MJ, et al: Thyroid disease and oral lichen planus as comorbidity. Dermatology 2016; 232: 214.
Garcia-Pola MJ, et al: Treatment of oral lichen planus. Med Clin (Barc) 2017; 149: 351.
Goettmann S, et al: Nail lichen planus. J Eur Acad Dermatol Venereol 2012; 26: 1304.
Goñi Esarte S, et al: Rituximab as rescue therapy in refractory esophageal lichen planus. Gastroentereol Hepatol 2013; 36: 264.
Gorouhi F, et al: Cutaneous and mucosal lichen planus: a comprehensive review of clinical subtypes, risk factors, diagnosis, and prognosis. ScientificWorldJournal 2014; 742826.
Gupta S, et al: Interventions for the management of oral lichen planus. Oral Dis 2017; 23: 1029.
Halonen P, et al: Cancer risk of lichen planus. Int J Cancer 2018; 142: 18.
Holló P, et al: Successful treatment of lichen planus with adalimumab. Acta Derm Venereol 2012; 92: 339.
Iraji F, et al: Comparison of the narrow band UVB versus systemic corticosteroids in the treatment of lichen planus. J Res Med Sci 2011; 16: 1578.
Ito Y, et al: Disseminated lichen planus due to a zinc allergy. J Dermatol 2012; 39: 948.
Jin X, et al: Association between -308 G/A polymorphism in TNF-α gene and lichen planus. J Dermatol Sci 2012; 68: 127.
Kanwar AJ, De D: Methotrexate for treatment of lichen planus. J Eur Acad Dermatol Venereol 2013; 27: e410.
Kern JS, et al: Esophageal involvement is frequent in lichen planus. Eur J Gastroenterol Hepatol 2016; 28: 1374.
Kurago ZB: Etiology and pathogenesis of oral lichen planus: an overview. Oral Surg Oral Med Oral Pathol Oral Radiol 2016; 122: 72.
Lade NR, et al: Blaschkoid lichen planus. Dermatol Online J 2013; 19: 17.
Le Cleach L, Chosidow O: Lichen planus. N Engl J Med 2012; 366: 723.
Lewis FM, Bogliatto F: Erosive vulval lichen planus. Eur J Obstet Gynecol Reprod Biol 2013; 171: 214.
Liakopoulou A, et al: Bullous lichen planus—a review. J Dermatol Case Rep 2017; 11: 1.
Limas C, Limas CJ: Lichen planus in children. Pediatr Dermatol 2002; 19: 204.
Lodi G, et al: Hepatitis C virus infection and lichen planus. Oral Dis 2010; 16: 601.
Lospinoso DJ, et al: Lupus erythematosus/lichen planus overlap syndrome. Lupus 2013; 22: 851.
Lucchese A, et al: Vulvovaginal gingival lichen planus. Oral Implantol (Rome) 2016; 9: 54.
Montebugnoli L, et al: Clinical and histologic healing of lichenoid oral lesions following amalgam removal. Oral Surg Oral Med Oral Pathol Oral Radiol 2012; 113: 766.
Morales-Callaghan A Jr, et al: Annular atrophic lichen planus. J Am Acad Dermatol 2005; 52: 906.
Muñoz ER, et al: Isolated conjunctival lichen planus. Arch Dermatol 2011; 147: 465.
Nagao Y, et al: Genome-wide association study identifies risk variants for lichen planus in patients with hepatitis C virus infection. Clin Gastroenterol Hepatol 2017; 15: 937.
Nakashima C, et al: Treatment of intractable oral lichen planus with intravenous immunoglobulin therapy. Eur J Dermatol 2012; 22: 693.
Nishizawa A, et al: Close association between metal allergy and nail lichen planus. J Eur Acad Dermatol 2013; 27: e231.
Pandhi D et al: Lichen planus in childhood. Pediatr Dermatol 2014, 31: 59.
Parodi A, et al: Prevalence of stratified epithelium-specific antinuclear antibodies in 138 patients with lichen planus. J Am Acad Dermatol 2007; 56: 974.
Passeron T, et al: Treatment of oral erosive lichen planus with 1% pimecrolimus cream. Arch Dermatol 2007; 143: 472.
Paul J, et al: An open-label pilot study of apremilast for the treatment of moderate to severe lichen planus. J Am Acad Dermatol 2013; 68: 255.
Payette MJ, et al: Lichen planus and other lichenoid dermatoses. Clin Dermatol 2015; 33: 631.
Quispel R, et al: High prevalence of esophageal involvement in lichen planus. Endoscopy 2009; 41: 187.
Radfar L, et al: A comparative treatment study of topical tacrolimus and clobetasol in oral lichen planus. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2008; 105: 187.
Rashed L, et al: Studying the association between methylenetetrahydrofolate reductase (MTHFR) 677 gene polymorphism, cardiovascular risk and lichen planus. J Oral Pathol Med 2017; 46: 1023.
Rasi A, et al: Efficacy of oral metronidazole in treatment of cutaneous and mucosal lichen planus. J Drugs Dermatol 2010; 9: 1186.
Rogers RS, Bruce AJ: Lichenoid contact stomatitis. Arch Dermatol 2004; 140: 1524.
Salgado DS, et al: Plaque control improves the painful symptoms of oral lichen planus gingival lesions. J Oral Pathol Med 2013; 42: 728.
Sartori-Valinotti JC, et al: A 10-year review of otic lichen planus. JAMA Dermatol 2013; 149: 1082.
Schmidgen MI, et al: Pembrolizumab-induced lichen planus pemphigoides in a patient with metastatic melanoma. J Dtsch Dermatol Ges 2017; 15: 742.
Scott GD, et al: New-onset cutaneous lichen planus following therapy for hepatitis C with ledipasvir-sofosbuvir. J Cutan Pathol 2016; 43: 408.
Shah KM, et al: Oral lichenoid reaction due to nickel alloy contact hypersensitivity. BMJ Case Rep 2013; 2013.
Simpson RC, et al: Real-life experience of managing vulval erosive lichen planus. Br J Dermatol 2012; 167: 85.
Singal A: Familial mucosal lichen planus in three successive generations. Int J Dermatol 2005; 44: 81.
Singh S, et al: Lichen planus causing severe vulval stenosis. Int J Dermatol 2013; 52: 1398.
Suresh SS, et al: Medical management of oral lichen planus. J Clin Diagn Res 2016; 10: ZE10-5.
Thorne JE, et al: Lichen planus and cicatrizing conjunctivitis. Am J Ophthalmol 2003; 136: 239.
Thornhill M, et al: The role of histopathological characteristic in distinguishing amalgam-associated oral lichenoid reactions and oral lichen planus. J Oral Pathol Med 2006; 35: 233.
Tiwari SM, et al: Dental patch testing in patients with undifferentiated oral lichen planus. Australas J Dermatol 2017; ePub ahead of print.
Wakade DV, et al: PD-1 inhibitors induced bullous lichen planus-like reactions. Melanoma Res 2016; 26: 421.
Walton KE, et al: Childhood lichen planus. Pediatr Dermatol 2010; 27: 34.
Webber NK, et al: Lacrimal canalicular duct scarring in patients with lichen planus. Arch Dermatol 2012; 148: 224.
Wee JS, et al: Efficacy of mycophenolate mofetil in severe mucocutaneous lichen planus. Br J Dermatol 2012; 167: 36.
Weston G, et al: Update on lichen planus and its clinical variants. Int J Womens Dermatol 2015; 1: 140.
Wilk M, et al: Annular lichenoid dermatitis (of youth). Am J Dermatopathol 2017; 39: 177.
Xia J, et al: Short-term clinical evaluation of intralesional triamcinolone acetonide injection for ulcerative oral lichen planus. J Oral Pathol Med 2006; 35: 327.
Yew YW, et al: A retrospective cohort study of epidemiology and clinical outcome in lichen planus. Ann Acad Med Singapore 2016; 45: 516.
Zaraa I, et al: Lichen planus pemphigoides. Int J Dermatol 2013; 52: 406.
Zendell K: Genital lichen planus. Semin Cutan Med Surg 2015; 34: 182.
Adnexal Lichen Planus: Follicular Lichen Planus (Lichen Planopilaris) and Acrosyringeal Lichen Planus
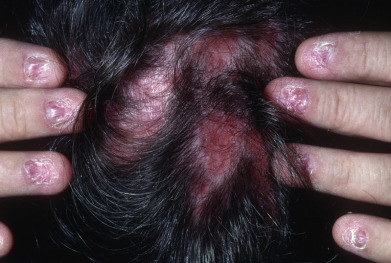
Lichen planopilaris (LPP) is lichen planus involving the follicular apparatus. Most cases involve the scalp, and LPP is an important cause of cicatricial alopecia ( Fig. 12.11 ; see Chapter 33 ). From 70%–90% or more of affected patients are women, usually about age 50. The oral mucosa may be involved in 7%–27% of patients, and 20%–40% of patients have cutaneous involvement. Patients often demonstrate perifollicular erythema and scale with progressive scarring; dermoscopy may reveal subtle signs of the folliculocentric inflammation. As with classic LP, there are reports of trauma inducing LPP. Patients may have lesions scattered throughout the scalp, clustered centrally, or on the margins; one study of 80 patients showed the FFA variant in 31% of patients with LPP. FFA is considered a variant of LPP markedly more common in postmenopausal women, with rare skin-colored papules on the upper face often lateral to the eyes, frequent loss of eyebrows, and occasionally loss of axillary and/or pubic hair. One study demonstrated an androgen excess in LPP and an androgen deficiency in patients with FFA. Graham-Little-Piccardi-Lasseur syndrome is a rare variant of LPP of the scalp with coexistent keratosis pilaris–like LPP lesions on the skin. Patients may rarely present with localized linear LPP. The rarest variant of LPP is lichen planus follicularis tumidus, formerly called agminate lichen follicularis with cysts and comedones. This presents in the retroauricular area and on the cheeks of middle-age women, where the lesions appear as tumid, red-violet plaques covered with numerous small, white-yellow cysts and comedones. The lesions resemble the plaques seen in Favre-Racouchot syndrome and phymatous cystic rosacea. The ears, chin, and scalp can be similarly involved. Other areas of LP of the skin and nails can occur in the same patient, and these may appear at about the same time. Histologically, a dense lichenoid infiltrate surrounds the follicles and cysts of the affected skin. The cysts are considered secondary to the lichenoid inflammation. Favre-Racouchot syndrome, follicular mucinosis, and LE must be distinguished histologically from LPP. LPP treatment often involves potent topical corticosteroids and/or intralesional injections, with systemic options including hydroxychloroquine, tetracycline-class antibiotics, retinoids, pioglitazones, and sometimes suppressive agents such as cyclosporine, mycophenolate, methotrexate, or systemic steroids. FFA has been treated with 5-α reductase inhibitors as well. Hair transplantation may be an option once the inflammation is controlled.