Disorder |
Inheritance |
Basic Defect |
Major Dermatologic Findings |
Associated Features |
Miscellaneous |
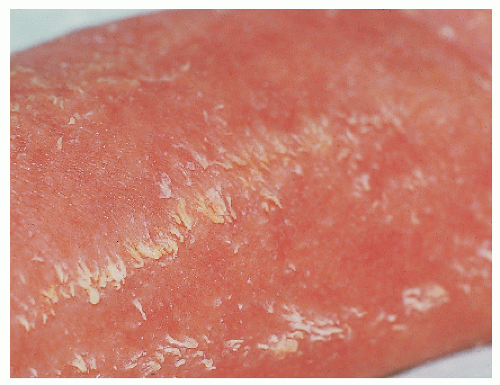
Ichthyosis vulgaris |
AD |
Unknown |
Mild-to-moderate white scales
Spares flexures and neck; involves face Keratosis pilaris Atopic dermatitis (50%) |
None |
Improves with age and warm weather |
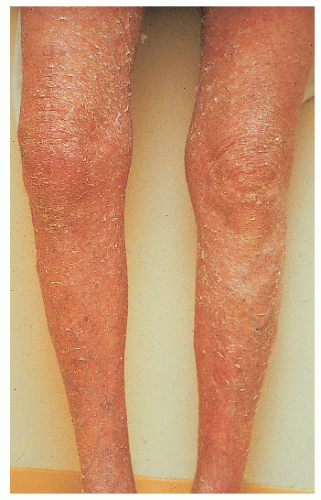
X-linked ichthyosis (sterol sulfatase deficiency) |
XLR |
Mutation/deletion of steroid sulfatase gene |
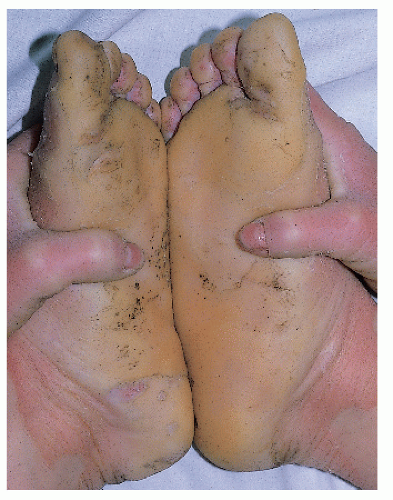
Moderate-to-severe white-brown scale Spares face; involves neck |
Corneal opacities Possible increased risk of testicular malignancy |
Pregnancies with affected males have low to absent estriol levels; failure of spontaneous initiation of labor is common |
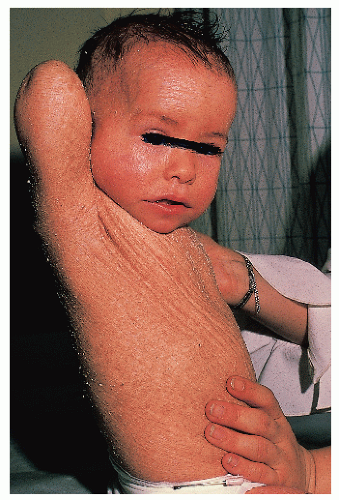
Bullous congenital ichthyosiform erythroderma (epidermolytic hyperkeratosis) |
AD |
Mutations in K1 or K10 (suprabasal keratins) |
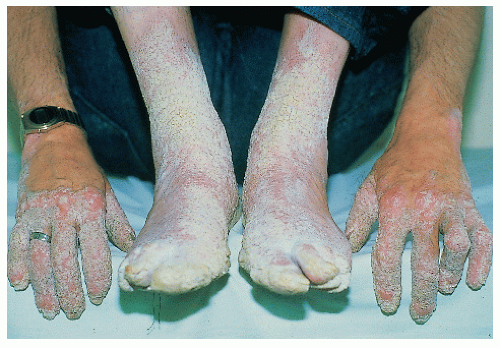
Red skin with blisters and scale evident at birth
Marked hyperkeratosis
Face usually least affected
Inter- and intrafamilial variability |
Secondary skin infection, bacterial and fungal common |
Skin is tender; skin fragility improves with age |
Lamellar ichthyosis/nonbullous congenital ichthyosiform erythroderma/congenital autosomal recessive ichthyosis |
AR |
Heterogenous. Some caused by mutations in transglutaminase 1 (TGM1), 12-R lipoxygenase (ALOX12B), lipoxygenase-3 (ALOXE3), ATP-binding cassette transporter 2 (ABCA2) |
LI: mild erythroderma; brown, adherent plate-like scale
NCIE: Erythroderma; fine, white scale
Many cases with overlap in phenotype |
Secondary tinea infection common |
Collodion membrane common at birth
Ectropion/ecla-bium common |
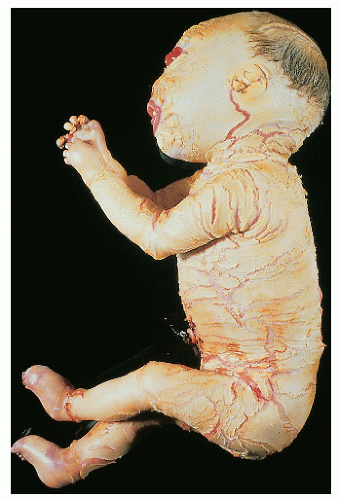
Harlequin fetus |
AR |
Unknown; probably heterogenous |
Severe, armor platelike hyperkeratosis In survivors, phenotype becomes similar to BCIE |
Among survivors, mental retardation has been noted in a few |
Rare spontaneous survival; handful of survivors treated with oral retinoids |
Conradi
Hunermann |
XLD
AR |
XLD: mutation in gene encoding delta(8)-delta(7) sterol isomerase emopamilbinding protein
AR: mutations in PEX7 gene |
Feathery scale on erythrodermic base
Follicular atrophoderma |
Seizures; MR
Chondrodysplasia punctata
Cataracts |
Asymmetry typical in XLD form |
Sjögren-Larsson syndrome |
AR |
Fatty aldehyde dehydrogenase deficiency |
Mild-to-moderate fine, adherent scale
Pruritus |
Progressive spastic paraparesis
Mild retardation
Glistening white dots on retina |
Netherton syndrome |
AR |
Mutations in SPINK5 gene |
Variable erythroderma and scale
Classic pattern of ichthyosis linearis circumflexa |
Trichorrhexis invaginata (bamboo hair) |
Failure to thrive
Food allergies |
Collodion baby |
AR if isolated Otherwise, depends on underlying disorder |
Heterogenous |
Plastic wrap-like membrane peels within few weeks after birth, revealing underlying skin, which may range from minimally xerotic to lamellar ichthyosis |
This is a feature of many disorders including lamellar ichthyosis, hypohidrotic ectodermal dysplasia, Gaucher disease and lamellar exfoliation of the newborn |
|
Abbreviations: AD, autosomal dominant; AR, autosomal recessive; BCIE, bullous congenital ichthyosiform erythroderma; XLR, X-linked recessive; K, keratin; MR, mental retardation. |