Flushing
Flushing presents with transient erythema, usually localized to the face, neck, and upper trunk, and a sensation of warmth. Flushing may be physiologic or pathologic; though normally benign, causes range from emotions to hormones to medications and malignancy.
Menopausal flushing may be associated with perspiration, as is flushing induced by high ambient temperature, fever, or consumption of alcohol or hot or spicy foods and beverages. Flushing associated with medications, histamine, or serotonin is generally dry. Menopausal flushing may be age related, may be induced by oophorectomy or medication (tamoxifen, leuprolide acetate, treptorelin, clomiphene citrate, danazol), and may begin long before menses cease. Paraxetine, other antidepressants, or gabapentin may help. Men may also experience climacteric flushing after orchiectomy or antiandrogen therapy (flutamide).
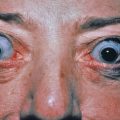
Flushing may occur through thermoregulation. Blushing, or emotional flushing, may be either emotionally or physiologically induced. Simple facial redness may occur in individuals with translucent skin and is called anatomically predisposed blushing. Intense flushing may be associated with rosacea. In patients with rosacea, exercise, ambient heat or cold, spicy foods, alcohol, and hot beverages are common triggers for flushing. Topical cinnamic aldehyde can induce flushing. Drugs associated with flushing include niacin, hormonal agents, serotonin agonists, calcium channel blockers, cyclosporine, chemotherapeutic agents, antimicrobials (vancomycin, metronidazole, rifampin), disulfiram, bromocriptine, intravenous contrast material, vasodilators (nicotinic acid, nitroglycerine, sildenafil and related drugs), and glucocorticoids. Severe serotonin toxicity with flushing can be precipitated by the combination of a monoamine oxidase inhibitor and a selective serotonin reuptake inhibitor (SSRI). Reduced or absent methylnicotinate-induced flushing has been noted in patients with schizophrenia. This lack of flushing in response to methylnicotinate has been used for diagnostic psychiatric testing. Flushing after induction of general anesthesia with agents such as thiopental and muscle relaxants is more common in patients prone to blushing. It appears to be neuronally mediated rather than related to histamine release. Endogenous vasoactive substances are associated with flushing in carcinoid syndrome, mastocytosis, medullary thyroid carcinoma, and pheochromocytoma.
Some food additives, such as nitrites, sulfites, and spicy foods with capsaicin, can induce flushing; monosodium glutamate (MSG) has been reported to as well, but remains controversial. Nitrites are found in deli meats and cured meats, and sulfites are found in wine, dried fruit, prepared foods, and fresh grapes and potatoes. Scombroid fish poisoning from improperly refrigerated fish presents with flushing and systemic symptoms within 10–30 minutes of eating. Alcohol may produce flushing in patients using topical calcineurin inhibitors. Individuals who flush without an identifiable cause should be investigated for dietary triggers and subtle manifestations of rosacea.
Many cases of flushing remain idiopathic. Atypical causes or mimics include superior vena cava syndrome, thyroid disease, and malignancies (mastocytosis, pheochromocytoma, carcinoid syndrome, and others). Urinary catecholamines and serotonin and histamine metabolites should be measured if an endogenous cause is suspected.
The Women’s Health Initiative studies concerning hormone replacement therapy (HRT) suggested that breast cancer risk is increased by combinations of estrogen and progestogen taken for longer than 5 years. Unopposed estrogen can increase the risk of endometrial carcinoma in premenopausal women. HRT does not appear to lower the risk of cardiac events, and the risks of long-term therapy often outweigh the benefits. Short-term HRT may still be very helpful in the management of perimenopausal flushing, because alternatives have generally been disappointing. Menopausal flushing may also respond to low-dose oral or transdermal estrogen. Flushing can be reduced by avoidance of alcohol, caffeine, and spicy foods. Niacin-induced flushing is mediated by prostaglandin D2 (PGD2). PGD2 shows some response to aspirin, as well as the PGD2 receptor-1 antagonist laropiprant.
Sadeghian A, et al: Etiologies and management of cutaneous flushing: malignant causes. J Am Acad Dermatol 2017; 77: 405.
Sadeghian A, et al: Etiologies and management of cutaneous flushing: nonmalignant causes. J Am Acad Dermatol 2017; 77: 391.
Erythemas
The term erythema means blanchable redness (hyperemia) of the skin. A number of reactive skin conditions are referred to as erythema. These include toxic erythemas related to viral and bacterial infections, erythema multiforme, erythema nodosum, and the gyrate (figurate) erythemas.
Erythema Palmare
Erythema palmare, or persistent palmar erythema, is usually most marked on the hypothenar areas and is associated with an elevated level of circulating estrogen. Cirrhosis, hepatic metastases, and pregnancy are common causes. Hereditary palmar erythema (Lane disease) has been rarely reported.
Generalized Erythema
Generalized erythema may be caused by medications, bacterial toxins, or viral infection. It is often uneven in distribution, being most noticeable on the chest, proximal extremities, and face. In general, these reactions are self-limited and resolve when the offending medication is stopped or the associated infection is treated or resolves. Specific exanthems associated with bacterial or viral infections are discussed in Chapters 14 and 19 .
Erythema Toxicum Neonatorum
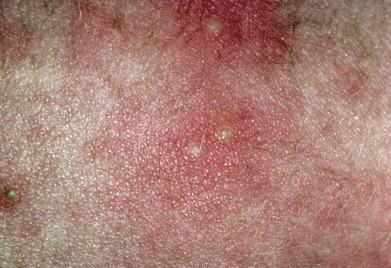
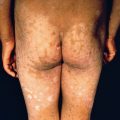
Erythema toxicum neonatorum occurs in a quarter to under half of healthy full-term newborns, usually on the second or third day of life. Because it is so common, dermatologists are usually consulted only for the most florid or atypical cases. Characteristically, the broad erythematous flare is much more prominent than the small follicular papule or pustule it surrounds ( Fig. 7.1 ). Lesions involve the face, trunk, and proximal extremities and appear rarely on the soles or palms. There may be confluent erythema on the face. Fever is absent, and the eruption generally disappears by the 10th day. Erythema toxicum must be distinguished from miliaria, bacterial folliculitis, neonatal herpes, and scabies. When the rash is atypical, smears of the pustules demonstrating eosinophils are adequate to confirm the diagnosis. Rarely, a biopsy is required and demonstrates folliculitis containing eosinophils and neutrophils.
Durieux-Verde M, et al: Erythema palmare hereditarium (Lane’s disease). Ann Dermatol Venereol 2016; 143: 32.
Reginatto FP, et al: Prevalence and characterization of neonatal skin disorders in the first 72h of life. J Pediatr 2017; 93: 238.
Erythema Multiforme
In 1860 von Hebra first described erythema exudativum multiforme. The original disease described by von Hebra is now called erythema multiforme minor (minus) or herpes simplex–associated erythema multiforme. It is strongly associated with a preceding herpetic infection. When multiple mucous membranes are involved, the lesions are more intense, and fever or arthralgias accompany the eruption, erythema multiforme major (majus) is diagnosed. This is most often caused by Mycoplasma infection. In contrast, Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) usually represent adverse reactions to medications (see Chapter 6 ). As treatment and prognosis are related in part to the inciting agent, it is useful to classify erythema multiforme (EM) as follows:
- •
Herpes simplex–associated EM (HAEM)
- •
Erythema multiforme major (most often caused by Mycoplasma; some suggest the term mycoplasma pneumonia-induced rash and mucositis )
- •
Chronic oral EM
- •
Contact dermatitis–induced EM (see Chapter 6 )
- •
Radiation-induced EM (see Chapter 6 )
- •
Idiopathic
Clinical Features
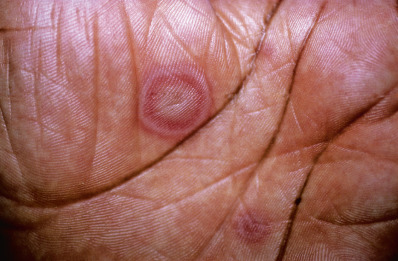
HAEM (erythema multiforme minor) is a recurrent self-limited disease, usually of young adults, occurring seasonally in the spring and fall, with each episode lasting 1–4 weeks. The individual clinical lesions begin as sharply marginated, erythematous macules, which become raised, edematous papules over 24–48 hours. The lesions may reach several centimeters in diameter. Typically, a ring of erythema forms around the periphery, and centrally the lesions become flatter, more purpuric, and dusky. This lesion is the classic “target” or “iris” lesion with three zones: central dusky purpura; an elevated, edematous, pale ring; and surrounding macular erythema ( Figs. 7.2 and 7.3 ). The central area may be bullous. Typical targets are best observed on the palms and soles. Lesions generally appear symmetrically and acrally, with initial involvement most frequently on the dorsal hands. The dorsal feet, extensor limbs, elbows, knees, palms, and soles typically become involved. In about 10% of patients, more widespread lesions occur on the trunk. The Koebner phenomenon or photoaccentuation may be observed. Mucosal involvement occurs in 25% of cases and is usually limited to the oral mucosa. Oral lesions may appear as indurated plaques, target lesions, or erosions ( Fig. 7.4 ).
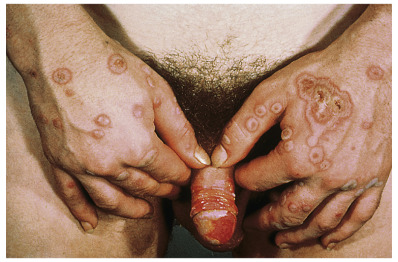
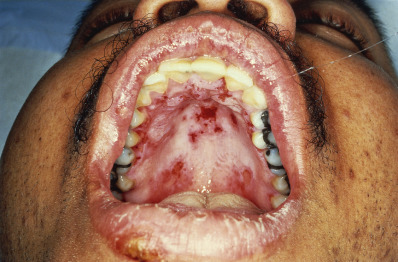
An atypical variant of HAEM has been described in women. It consists of outbreaks of unilateral or segmental papules and plaques that may be few in number or solitary. Lesions may be up to 20 cm in diameter. The plaques are erythematous and evolve to have a dusky center, which desquamates. Subcutaneous nodules resembling erythema nodosum may be simultaneously present. Histologic examination shows features of EM, and herpes simplex virus (HSV) DNA may be identified in the lesions by polymerase chain reaction (PCR). Acyclovir suppression prevents the lesions.
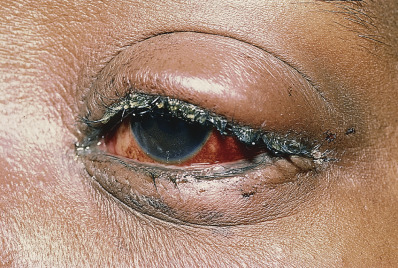
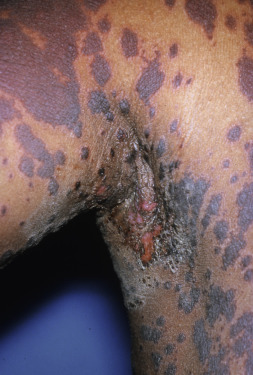
Erythema multiforme major is frequently accompanied by a febrile prodrome and sometimes arthralgias. It occurs in all ages, is centered on the extremities and face, but more often than EM minor may include truncal lesions, which are papular and erythematous to dusky in color. Mucous membrane disease is prominent and often severely involves the oral mucosa and lips with hemorrhagic sloughing; less commonly the genital and ocular mucosa may be involved as well ( Fig. 7.5 ). SJS is distinguished morphologically by the presence of purpura or bullae in macular lesions of the trunk ( Fig. 7.6 ). In children, polycyclic urticarial lesions often become dusky centrally and are frequently misdiagnosed as EM. This presentation of urticaria has been dubbed “urticaria multiforme.” It represents urticaria, and histologic changes of EM are never present.
Etiologic Factors
Typical EM minor is usually associated with a preceding orolabial HSV infection. HAEM lesions appear 1–3 weeks (average 10 days) after the herpes outbreak. Episodes of EM minor may not follow every episode of herpes, and some EM outbreaks will not be preceded by a clinically recognizable herpetic lesion. Using PCR and in situ hybridization techniques, HSV DNA and antigens have been found in the lesions of EM minor. The majority of “idiopathic” cases of EM minor are associated with recurrent HSV infection, and patients may be successfully treated with suppressive antiviral regimens. Autoantibodies have been reported to a number of desmosomal adherence molecules, but whether these are pathogenic or incidental (due to epitope spreading from repeated exposure) is unclear. EM major is associated with Mycoplasma infections, although a minority may result from herpes simplex and a reaction to medications.
Histopathology
The histologic features are similar in HAEM and EM major and are not predictive of etiology. The extent of epidermal involvement depends on the duration of the lesion and where in the lesion the biopsy is taken. All lesions are characterized by cellular necrosis. Biopsies of EM demonstrate a normal basket-weave stratum corneum and a vacuolar interface reaction. Vacuoles and foci of individual cell necrosis are present and out of proportion to the number of lymphocytes. The dermal infiltrate is largely mononuclear and tends to be primarily around the upper dermal vessels and along the dermoepidermal junction. Activated T lymphocytes are present in lesions of EM, with cytotoxic or suppressor cells more prominent in the epidermis and helper T cells in the dermis. Leukocytoclastic vasculitis is not observed. Eosinophils may be present but are rarely prominent. The presence of eosinophils is not predictive of the etiology. Histologically, EM must be distinguished from the following:
- •
Fixed drug eruption, which often has a deeper infiltrate, eosinophils and neutrophils, papillary dermal fibrosis, and melanophages
- •
Graft-versus-host disease (GVHD), which typically has a more compact stratum corneum and involves the follicles
- •
Pityriasis lichenoides, which characteristically has a lymphocyte in every vacuole, erythrocyte extravasation, and neutrophil margination within dermal vessels
- •
Lupus erythematosus, which has compact hyperkeratosis, a deeper periadnexal infiltrate, dermal mucin, and basement membrane zone thickening
Differential Diagnosis
When characteristic target lesions are present, the diagnosis of EM is established clinically. When bullae are present, EM major must be distinguished from bullous arthropod reactions and autoimmune bullous diseases: pemphigus if mucous membrane involvement is prominent, and bullous pemphigoid if lesions are small and erythema is prominent at the periphery of the bulla. Paraneoplastic pemphigus may produce atypical target lesions, mucosal involvement, and a vacuolar interface dermatitis and may appear similar to Mycoplasma -induced EM major. Use of direct immunofluorescence may be necessary to exclude this possibility.
Treatment
Treatment of EM is determined by its cause and extent. EM minor is generally related to HSV, and prevention of herpetic outbreaks is central to control of the subsequent episodes of EM. A sunscreen lotion and sunscreen-containing lip balm should be used daily on the face and lips to prevent ultraviolet B (UVB)–induced outbreaks of HSV. If this does not prevent recurrence or if genital HSV is the cause, chronic suppressive doses of an oral antiviral drug (valacyclovir, 500 mg to 1 g/day, or famciclovir) may be used. If this dose is ineffective, it may be increased. This will prevent recurrences in up to 90% of HSV-related cases. Intermittent treatment with systemic antivirals or the use of topical antivirals is of minimal benefit in preventing HAEM. It should be noted that most cases of EM minor (HAEM) are self-limited, and symptomatic treatment may be all that is required. Symptoms related to oral lesions often respond to topical “swish and spit” mixtures containing lidocaine, diphenhydramine (Benadryl), and kaolin. In extensive cases of EM minor, intermittent steroids, or chronic dapsone, cyclosporine, azathioprine, or thalidomide may occasionally be helpful. Apremilast and rituximab have recently been reported for recalcitrant cases. If HSV infection is present, concurrent antivirals should be used. For patients with widespread EM unresponsive to the previous therapies, management is as for severe drug-induced SJS (see Chapter 6 ).
Oral Erythema Multiforme
A unique subset of EM is limited to or most prominent in the oral cavity. Clinically, patients are otherwise well; 60% are female, with a mean age of 43 years. The minority, about 25%, have recurrent, self-limited, cyclic disease. The oral cavity is the only site of involvement in 45%, in 30% there is oral and lip involvement, and in 25% the skin is also involved. All portions of the oral cavity may be involved, but the tongue, gingiva, and buccal mucosa are usually most severely affected. Lesions are almost universally eroded, with or without a pseudomembrane. There are no well-designed trials of treatment for oral EM, but the treatments previously listed for EM minor are typically used; topical corticosteroids may be helpful (fluocinonide gel 0.05%). “Swish and spit” mixtures containing lidocaine, diphenhydramine, and kaolin are helpful for symptomatic relief; patients should be warned the anesthetic effect may dampen their gag reflex.
Canavan TN, et al: Mycoplasma pneumonia-induced rash and mucositis as a syndrome distinct from Stevens-Johnson syndrome and erythema multiforme. J Am Acad Dermatol 2015; 72: 239.
Chen T, et al: Apremilast for treatment of recurrent erythema multiforme. Dermatol Online J 2017 Jan 15; 23.
Ellis E, et al: Circulating plakin autoantibodies in a patient with erythema multiforme major. Australas J Dermatol 2014; 55: 266.
Hirsch G, et al: Rituximab, a new treatment for difficult-to-treat chronic erythema multiforme major? J Eur Acad Dermatol Venereol 2016; 30: 1140.
Oak AS, et al: Treatment of antiviral-resistant recurrent erythema multiforme with dapsone. Dermatol Ther 2017; 30.
Roujeau JC, et al: Re-evaluation of ‘drug-induced’ erythema multiforme in the medical literature. Br J Dermatol 2016; 175: 650.
Siedner-Weintraub Y, et al: Paediatric erythema multiforme. Acta Derm Venereol 2017; 97: 489.
Gyrate Erythemas (Figurate Erythemas)
The gyrate erythemas are characterized by clinical lesions that are round (circinate), ringlike (annular), polycyclic (figurate), or arcuate. The primary lesions are erythematous and slightly elevated. There may be a trailing scale, as in erythema annulare centrifugum. In some of these diseases, the lesions are transient and migratory, and in some they are fixed. Gyrate erythemas often represent the cutaneous manifestations of an infection, malignancy, or drug reaction. Certain diseases in this group have specific causes (erythema marginatum of rheumatic fever, carrier state of chronic granulomatous disease, erythema migrans of Lyme borreliosis) and are discussed in the relevant chapters.
Erythema Annulare Centrifugum
Erythema annulare centrifugum (EAC) is the most common gyrate erythema. It is characterized by asymptomatic annular or polycyclic lesions that grow slowly (2–3 mm/day), rarely reaching more than 10 cm in diameter. Characteristically, there is a trailing scale at the inner border of the annular erythema ( Fig. 7.7 ). The surface is typically devoid of crusts or vesicles, although atypical cases with telangiectasia and purpura have been described. Lesions usually occur on the trunk and proximal extremities. Mucosal lesions are absent.
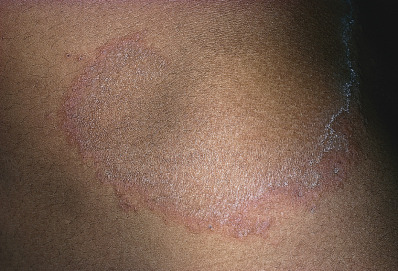
Histologically, the epidermis will show mild focal spongiosis and parakeratosis. Within the superficial dermis and at times the deep dermis, lymphocytes are organized tightly around the blood vessels in a pattern described as a “coat sleeve” arrangement. The gyrate erythemas are divided into the superficial and deep types, but these histologic types do not correlate with etiology.
Waxing and waning in severity, EAC tends to be recurrent over months to years. Most cases eventually subside spontaneously. While active, the eruption is often responsive to topical steroids. Topical calcipotriol has also been reported to be successful.
The majority of EAC cases are idiopathic. Some cases are clearly associated with dermatophytosis or the ingestion of molds, such as those in blue cheese. Other foods, such as tomatoes, are sometimes implicated, and a dietary journal may be helpful. Medications are implicated in some cases, and internal cancer has been found (solid organ and hematologic). Annually recurrent EAC has been reported. Laboratory tests should be dictated by the physical examination and associated signs and symptoms. In one study of 66 patients, 48% had an associated cutaneous fungal infection such as tinea pedis, and 13% had internal malignancies.
The differential diagnosis of EAC includes conditions that can have annular configuration, including granuloma annulare, secondary syphilis, tinea, subacute cutaneous lupus erythematosus, sarcoidosis, Hansen disease, erythema marginatum, erythema migrans, annular urticaria, and mycosis fungoides. Histologic examination, clinical features, and basic laboratory examinations will usually allow these diseases to be excluded.
Erythema Gyratum Repens
Erythema gyratum repens (EGR) is a rare disease that is striking and unique in appearance. Lesions consist of undulating wavy bands of slightly elevated erythema with trailing scale over the entire body. Lesions migrate rapidly (up to 1 cm/day) and are characteristically concentric, giving the skin a “wood grain” appearance ( Fig. 7.8 ).
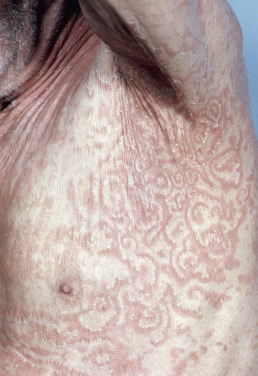
Pruritus may be severe, and blood eosinophilia is often found. In more than 70% of patients, an underlying malignancy is found. Lung cancer is the most common associated malignancy, although a wide range of neoplasms has been described. The skin eruption precedes the detection of the malignancy by an average of 9 months. Given the high frequency of malignant disease, patients with EGR should have extensive evaluations to exclude internal malignancy. If the carcinoma is removed, the lesions clear. Otherwise, the eruption is generally resistant to treatment, although cetirizine and topical corticosteroids have been reported to improve individual cases. EGR may rarely occur due to other causes, including medications, infections (pulmonary tuberculosis, cryptogenic organizing pneumonia), or other systemic diseases (rheumatoid arthritis). These patients respond to discontinuation of the implicated medication or to treatment of the underlying condition.
Eosinophilic Annular Erythema
Adults or children may develop bilateral annular erythema, usually presenting on the trunk and often symmetric. Females are the favored sex. Histologically a dense perivascular and interstitial lymphocytic infiltrate with many eosinophils is seen but without flame figures. Spontaneous resolution may occur. Hydroxychloroquine and/or prednisone are favored treatments when needed, though phototherapy, nicotinamide, and other treatments have been used. Normally an isolated occurrence, there are reports in the setting of thymoma, prostate cancer, and autoimmune pancreatitis.
Neutrophilic figurate erythema of infancy is a variant with a dermal neutrophilic infiltrate and karyorrhexis on biopsy.
Abarzua A, et al: Eosinophilic annular erythema in childhood. An Bras Dermatol 2016; 91: 503.
Endo Y, et al: Erythema gyratum repens preceding the onset of rheumatoid arthritis. Eur J Dermatol 2013; 23: 399.
Mandel VD, et al: Annually recurring erythema annulare centrifugum. J Med Case Rep 2015; 9: 236.
Mu EW, et al: Paraneoplastic erythema annulare centrifugum eruption (PEACE). Dermatol Online J 2015 Dec 16; 21.
Ogawa K, et al: Eosinophilic annular erythema in a patient with autoimmune pancreatitis. J Dermatol 2016; 43: 1380.
Samotij D, et al: Erythema gyratum repens associated with cryptogenic organizing pneumonia. Indian J Dermatol Venereol Leprol 2016; 82: 212.
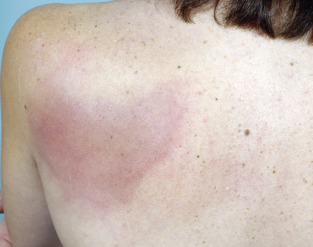
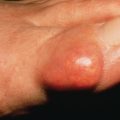
Eosinophilic Cellulitis (Wells Syndrome)
In 1971 Wells described four patients with acute onset of plaques resembling cellulitis that persisted for many weeks ( Fig. 7.9 ). Wells syndrome occurs at all ages, and pruritus is common. The condition is typically recurrent, and individual episodes may be prolonged. Degranulation of dermal eosinophils produces the flame figures seen in histologic sections. These consist of dermal collagen with adherent eosinophil granules. Eosinophilic panniculitis may also be present.
It is unclear whether Wells syndrome is a distinct disorder sui generis or a reaction pattern to many possible allergic stimuli. Many (perhaps most) cases represent arthropod reactions. It has also been associated with onchocerciasis, intestinal parasites, varicella, mumps, immunization, drug reactions that include interferon (IFN) and the anti–tumor necrosis factor (TNF)–α biologic agents, hypereosinophilic syndrome, myeloproliferative diseases, angioimmunoblastic lymphadenopathy, solid organ malignancies, atopic diathesis, inflammatory bowel disease (IBD), eosinophilic granulomatosis with polyangiitis (EGPA, Churg-Strauss syndrome), and fungal infection. Reports of Wells in association with chronic lymphocytic leukemia may represent another form of exuberant arthropod response in that disease. Most cases resolve without therapy; treatment may include topical and intralesional corticosteroids, oral antihistamines, or tacrolimus ointment. Systemic therapy for severe cases may include doxycycline, minocycline, phototherapy, antimalarials, dapsone, colchicine, sulfasalazine, low-dose prednisone, azathioprine, cyclosporine. Despite that some cases may be caused by exposure to TNF inhibitors, adalimumab has been used as well. Any underlying disease or triggering factor, including arthropod bites, should be eliminated.
Kambayashi Y, et al: Eosinophilic cellulitis induced by subcutaneous administration of interferon-β. Acta Derm Venereol 2013; 93: 755.
Qiao J, et al: Flame figures associated with eosinophilic dermatosis of hematologic malignancy. Int J Clin Exp Pathol 2013; 6: 1683.
Rabler F, et al: Treatment of eosinophilic cellulitis (Wells syndrome). J Eur Acad Dermatol Venereol 2016; 30: 1465.
Rajpara A, et al: Recurrent paraneoplastic Wells syndrome in a patient with metastatic renal cancer. Dermatol Online J 2014 Jun 15; 20.
Simpson JK, et al: Influenza vaccination as a novel trigger of Wells syndrome in a child. Pediatr Dermatol 2015; 32: e171.
Stuhr PM, et al: Wells syndrome associated with chronic lymphocytic leukemia. An Bras Dermatol 2015; 90: 571.
Reactive Neutrophilic Dermatoses
As with the gyrate erythemas, the reactive neutrophilic dermatoses tend to follow certain stimuli, such as acute upper respiratory tract infections (URIs) or medications, or are associated with underlying diseases, such as IBD and hematologic malignancy. Some of the neutrophilic dermatoses share common triggers, and clinical features may overlap. Patients may exhibit the simultaneous or sequential appearance of two or more of the conditions. Most often is the combination of typical lesions of Sweet syndrome on the upper body and erythema nodosum (EN)–like lesions on the legs. In these patients, histology often enables the diagnosis of subcutaneous Sweet syndrome for the EN-type lesions, allowing one diagnosis to be made. In occasional cases, however, it may be difficult to establish the diagnosis firmly as one of the neutrophilic reactive dermatoses. For these reasons, it is clinically useful to think of these diseases as forming a spectrum of conditions expressed in certain individuals by a group of stimuli with various overlapping morphologies.
Erythema Nodosum
Erythema nodosum is discussed in Chapter 23 .
Sweet Syndrome (Acute Febrile Neutrophilic Dermatosis)
Since its first description in 1964 by Dr. Robert Sweet, as a recurrent febrile dermatosis in women, the spectrum of this syndrome has expanded. Sweet syndrome primarily affects adults, and females outnumber males (reports vary with ranges from 3 : 1 up to 8 : 1 predominance, though some recent reports demonstrate near equivalent sex distribution). In younger adults, female predominance is marked, but in persons older than 50, the gender ratio is more equal, and cases associated with malignancy have a 1 : 1 ratio. In children, boys and girls are equally affected. In Europe, cases are more common in the spring and fall. Four main subtypes of Sweet syndrome have been described, based on their pathogenesis: the classic type (majority of cases), cases associated with neoplasia (3%–35% of cases), cases associated with inflammatory disease (3%–16%), and cases associated with pregnancy (1%–4%); drug-induced cases exist as well (1%–26%).
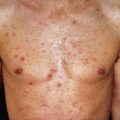
The clinical features of all four subtypes are similar, although dusky bullous lesions that overlap with pyoderma gangrenosum (PG) are more common in patients with associated leukemia. The primary skin lesion is a sharply marginated, rapidly extending, tender, erythematous or violaceous, painful, elevated plaque, 2–10 cm in diameter. Lesions may appear intensely edematous (“juicy”), or merely indurated ( Fig. 7.10 ). They typically involve the face ( Fig. 7.11 ), neck, upper trunk, and extremities. Lesions may burn but do not itch. The surface of the plaques may develop pseudovesiculation or pustulation as a result of an intense dermal inflammatory infiltrate and accompanying dermal edema. Pathergy or koebnerization may occur in 25%–30% of patients, from trauma, biopsies, peripheral IV lines, and rarely from phototherapy or radiation. Clinical morphologic variants more recently described include immunocompromised patients with deep necrosis simulating necrotizing fasciitis and patients whose lesions were extremely large and described as giant cellulitis–like in appearance.
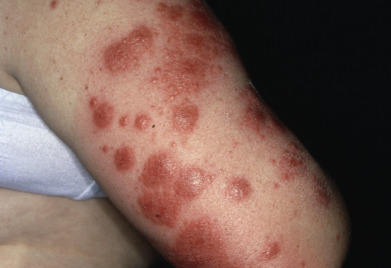
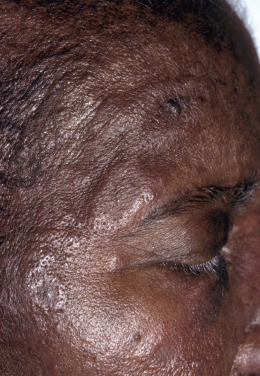
More than three quarters of Sweet syndrome patients have systemic findings. The most common is fever, present in 50%–80% of patients. Arthritis, arthralgias, or myalgias occur in one third to two thirds of cases. About 30% of patients have conjunctivitis or episcleritis. Other ocular manifestations include periorbital inflammation, dacryoadenitis, limbal nodules, peripheral ulcerative keratitis, glaucoma, iritis, and choroiditis. Oral lesions resembling aphthae occur in 2% or 3% of classic cases, but in 10% or more of those associated with hematologic malignancy. Cough, dyspnea, and pleuritis may represent pulmonary involvement. Pulmonary infiltrates and effusions are often seen on chest radiographs of such patients. Rarely, there may be cardiac, renal, hepatic, intestinal, and neurologic involvement. Multifocal sterile osteomyelitis may occur.
Laboratory findings include an elevated sedimentation rate (90%), neutrophilia (70%), leukocytosis (60%), and a left shift (increased bands; 50%). Patients with complete blood count (CBC) abnormalities may be more likely to have an underlying malignancy. Antineutrophilic cytoplasmic antibodies (ANCAs) have been rarely reported. In most cases, an attack lasts 3–6 weeks and then resolves. Recurrences may be seen with the same precipitating cause, such as URI. Persistent cases, with new lesions erupting before the old lesions resolve, may continue for many years.
The histologic hallmark of Sweet syndrome is a nodular and diffuse dermal infiltrate of neutrophils with karyorrhexis and massive papillary dermal edema. Leukocytoclastic vasculitis may be present focally, and this does not exclude a diagnosis of Sweet syndrome. Upper dermal edema may be so intense as to form subepidermal bullae. Leukemic cells may be present in the infiltrate, and clonal restriction of neutrophils has even been seen in Sweet syndrome not associated with malignancy.
Histologic variants described as histiocytic or lymphocytic Sweet syndrome have been reported. These entities are somewhat controversial, as Sweet syndrome is by definition a neutrophilic dermatosis. Occasionally, the main infiltrating cell resembles histiocytes, but in some studies with immunohistochemical stains, they are found to be immature myeloid cells, with positive myeloperoxidase stains. These can be mistaken for, and must be distinguished from, leukemia cutis. Patients with histiocytoid Sweets are more likely to have an underlying hematologic disorder or malignancy concurrent or shortly after the diagnosis is made.
The majority of cases of Sweet syndrome follow a URI or viral gastroenteritis and are therefore acute and self-limited. Other associated conditions include infections with Yersinia, toxoplasmosis, histoplasmosis, salmonellosis, tuberculosis, tonsillitis, vaccination, and vulvovaginal infections. Sweet syndrome has been reported in association with IBD and Behçet syndrome, and can develop in sites of lymphedema.
Hematologic malignancies or solid tumors are present in about 10% (3%–35% range) of reported cases. Sweet syndrome often presents early in the course of the cancer, but may present months to years before a diagnosis of malignancy is made. Sweet syndrome may occur with myelodysplastic syndromes or with hemoproliferative malignancies including leukemias (usually acute myelogenous) and lymphomas. One study reported an increased frequency of FLT3 mutations or del(5q) karyotype in AML patients with Sweets. Solid tumors are of any type but are most often genitourinary, breast (in women), or gastrointestinal (in men). Anemia is found in 93% of men and 71% of women with malignancy-associated Sweet syndrome. Thrombocytopenia is seen in half. Solitary, bullous, or ulcerative lesions are more frequently associated with malignancy, as is the histiocytoid histopathologic pattern.
Pregnancy-associated Sweet syndrome typically presents in the first or second trimester with lesions on the head, neck, trunk, and less often on the upper extremities. Lower-extremity lesions resembling EN may occur. The condition may resolve spontaneously or clear with topical or systemic corticosteroids. It may recur with subsequent pregnancies, but there seems to be no risk to the fetus.
Many drug therapies have been associated with Sweet-like reactions in the skin. The strongest association exists for granulocyte colony-stimulating factor (G-CSF). All- trans -retinoic acid (ATRA) and the new class of FLT3-inhibitors, which also have been reported to cause sweets, are, like G-CSF, often used in the treatment of diseases (such as leukemia) that themselves can be associated with Sweets, making causality assessments a challenge. Oral contraceptives, radiation therapy fields, radiocontrast dye, IFNs, interleukin-2 (IL-2), abatacept, TNF inhibitors, allopurinol, vaccines, multiple antineoplastic agents (bortezomib, gemcitabine, BRAF inhibitors, multikinase inhibitors, immunotherapy), anticonvulsants, antimicrobials (trimethoprim-sulfamethoxazole, minocycline, and many others), and azathioprine have been implicated. ATRA and FLT3 inhibitors cause terminal differentiation of some leukemic clones and are used to treat promyelocytic leukemia. After about 2 weeks of treatment, Sweet-like lesions may appear. Induction of the skin lesions appears to be related to the desired pharmacologic effect of the medication. ATRA has been reported to cause scrotal ulcers with neutrophils on histopathology; this is likely a site-specific manifestation of Sweet syndrome.
The two major criteria for the diagnosis of Sweet syndrome are the presence of red edematous plaques and a biopsy demonstrating neutrophils, karyorrhexis, and marked papillary dermal edema. Minor criteria include associated symptoms or conditions, laboratory findings, and response to therapy. Patients should have both major criteria and two of the four minor criteria for diagnosis ( Box 7.1 ). Sweet syndrome is a diagnosis of exclusion, and often tissue culture is necessary to exclude infection. Bowel bypass syndrome has skin lesions that, on histologic examination, are identical to those of Sweet syndrome; fever and arthritis also accompany bowel bypass syndrome. Although it is easy to distinguish classic EN from Sweet syndrome, these two conditions share many features. Both occur most often in young adult women and frequently follow URIs. Both may be associated with pregnancy, underlying malignancy, and IBD. In both, fever and arthritis may occur, along with leukocytosis with neutrophilia. There are many reports of simultaneous or sequential EN and Sweet syndrome in the same patient. Leukemia-associated Sweet syndrome may overlap with pyoderma gangrenosum. A search for an underlying cause should be undertaken, especially in persons over age 50 and those with anemia, thrombocytopenia, histiocytoid pathology, or lesions that are bullous or necrotic. The standard treatment is systemic corticosteroids, with approximately 1–2 mg/kg/day of oral prednisone. This will result in resolution of fever and skin lesions within days. Topical or intralesional steroids may be tried for mild disease but are often ineffective. Dapsone, colchicine, potassium iodide (which may rarely cause paradoxic worsening similar to iododerma), sulfapyridine, doxycycline, clofazimine, and nonsteroidal antiinflammatory drugs (NSAIDs) may be helpful in chronic or refractory disease. Granulocyte adsorption apheresis, cyclosporine, intravenous immune globulin (IVIG), thalidomide/lenalidomide, methotrexate, rituximab, TNF inhibitors, and anakinra have been used for severe cases. Medication should be continued for several weeks to prevent relapse.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


