The skin interacts with the endocrine system in many ways. Some of these are discussed in this chapter.
Leventhal JS, et al: Skin manifestations of endocrine and neuroendocrine tumors. Semin Oncol 2016; 43: 335.
Quatrano NA, et al: Dermatologic manifestations of endocrine disorders. Curr Opin Pediatr 2012; 24: 487.
Acromegaly
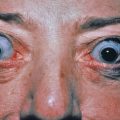
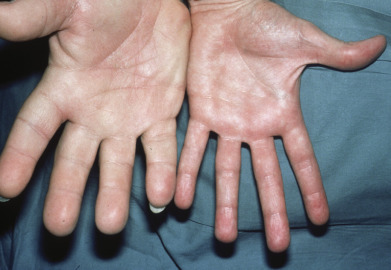
Excess growth hormone (GH) in prepubertal children leads to gigantism, whereas once the epiphyseal growth plates close, such excess leads to acromegaly. In acromegaly, changes in the soft tissues and bones form a characteristic syndrome. In association with the well-known changes in the facial features caused by gigantic hypertrophy of the chin, nose, and supraorbital ridges, there is thickening, reddening, and wrinkling of the forehead and exaggeration of the nasolabial grooves. The lips and tongue are thick. Cutis verticis gyrata is present in approximately 30% of patients. The hands and feet enlarge ( Fig. 24.1 ), and there is gradual growth of the fingertips until they resemble drumsticks. There is diffuse hypertrophy of the skin, which is at least partly caused by deposition of colloidal iron-positive material in the papillae and reticular dermis. This increased skin thickness can be demonstrated in lateral radiographs of the heel, with reversal toward normal after treatment. Skin thickness does not correlate well with GH levels at the time of diagnosis. Skin tags are often present and the skin has an oily feel. Hypertrichosis, hyperpigmentation, and hyperhidrosis occur in many patients. The viscera also enlarge and patients may develop a variety of rheumatologic, cardiovascular, metabolic, and respiratory complications.
The clinical changes may suggest the leonine facies of Hansen disease, as well as Paget disease, myxedema, and pachydermoperiostosis. Acromegaloid facial appearance syndrome is an inherited condition in which only the facial changes are present, and no abnormality of GH exists. Pseudoacromegaly is an acquired condition that may be seen in patients with severe insulin-resistant diabetes, which appears to be a fibroblast defect, in patients receiving long-term minoxidil.
The cause of 98% of acromegaly is hypersecretion of GH by a pituitary adenoma. Rare cases of ectopic GH-releasing hormone (GHRH) producing tumors of the lung and pancreas have been reported. The peak age of diagnosis is in the forties. Measurement of serum insulin-like growth factor (IGF, somatomedin C), measurement of serum GH after a glucose load, and magnetic resonance imaging (MRI) of the pituitary are diagnostic tests. It may occur as one of the manifestations of Carney complex, McCune-Albright syndrome, or multiple endocrine neoplasia (MEN) type I.
The currently preferred treatment is a transsphenoidal microsurgical excision of the tumor. Medical therapy may be used as a primary treatment for those unsuitable for surgery, as a preoperative treatment, or as secondary therapy after failed surgery. Octreotide and lanreotide are potent, long-acting inhibitors of GH (somatostatin analogs) that are given as once-monthly or biweekly intramuscular (IM) depot injections. Fatigue, paresthesias, and headaches improve rapidly. With continuous treatment, soft tissue swelling and facial coarsening improve as GH levels decline in almost all patients. After 18–24 months of therapy, 50% of patients will completely normalize, with the exception of hyperhidrosis, which persists in most patients. The dopamine agonists bromocriptine and cabergoline suppress GH secretion and are used as an adjuvant medical therapy in some cases. The GH receptor antagonist pegvisomant is another medical option to normalize GH secretion. Radiation is generally reserved for recalcitrant cases.
Akoglu G, et al: Cutaneous findings in patients with acromegaly. Acta Dermatovenereol Croat 2013; 21: 224.
Borson-Chazot F, et al: Acromegaly induced by ectopic secretion of GHRH. Ann Endocrinol 2012; 73: 497.
Chakraborty PP, et al: Pseudoacromegaly in congenital generalized lipodystrophy (Berardinelli-Seip syndrome). BMJ Case Rep 2016 Apr 11; 2016.
Davidovici BB, et al: Cutaneous manifestations of pituitary gland diseases. Clin Dermatol 2008; 26: 288.
Ghazi A, et al: Acromegaloid facial appearance. Case Rep Endocrinol 2013; 2013: 970396.
Jallad RS, et al: The place of medical treatment of acromegaly. Expert Opin Pharmacother 2013; 14: 1001.
Ribeiro-Oliveira A, et al: The changing face of acromegaly: advances in diagnosis and treatment. Nat Rev 2012; 8: 605.
Cushing Syndrome
Chronic excess of glucocorticoids leads to a wide variety of signs and symptoms. The most prominent features of Cushing syndrome include central obesity, affecting the face, neck, trunk, and especially the abdomen, but sparing the limbs. There is classically deposition of fat over the upper back, referred to as a buffalo hump. This may be treated with liposuction. The face becomes moon shaped, being wide and round. The peak age of onset is in the twenties and thirties.
The striking and distressing skin changes include hypertrichosis, dryness, acne, susceptibility to superficial dermatophyte and Pityrosporon infections, a plethora over the cheeks, anterior neck, and V of the chest, and the characteristic purplish, atrophic striae that may involve the abdomen ( Fig. 24.2 ), buttocks, back, breasts, upper arms, and thighs. Skin fragility and thinning occur such that easy bruising and a cigarette paper–type wrinkling are present. The skin may easily pull off when adhesive tape is removed (Liddle sign). The thinning of the skin can be demonstrated and measured in lateral radiographs of the heels. There is reversal with treatment. Women, who are affected four times more frequently than men in noniatrogenic cases, develop facial lanugo hypertrichosis, with thinning of the scalp hair. Occasionally, there may be livedo reticularis, purpura, ecchymosis, or brownish pigmentation. Poikiloderma-like changes have been observed. Opportunistic fungal infections occur, either with organisms that are not normally pathogenic or as uncommon presentations of common infections.
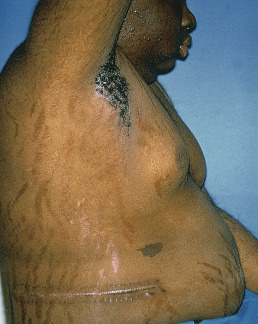
Patients with Cushing syndrome usually have hypertension and marked generalized arteriosclerosis, with progressive weakness, prostration, and pains in the back, limbs, and abdomen; kyphosis of the dorsal spine also occurs, accentuating the buffalo hump appearance. Osteoporosis occurs, and there is generally a loss of libido. In 20% of patients, a disturbance in carbohydrate metabolism develops, with hyperglycemia, glycosuria, and diabetes mellitus.
These varied symptoms indicate a marked and widespread disturbance caused by the hyperactive adrenal cortex. When microadenomas of the pituitary gland produce these clinical findings, it is referred to as Cushing disease; this accounts for only 10% of patients. Between 40% and 60% of additional cases are caused by increased adrenocorticotropic hormone (ACTH, corticotropin) production by the pituitary, but no adenoma is identified. Adrenal adenomas and carcinomas, with ectopic production of ACTH by other tumors, account for the remainder of cases of noniatrogenic Cushing syndrome. Iatrogenic Cushing syndrome is usually secondary to systemic administration of corticosteroids; however, absorption from topical corticosteroids to the skin, nasal mucosa, the conjunctiva, or the gingiva may occur, especially in children. Primary pigmented nodular adrenocortical disease leading to Cushing syndrome occurs in 30% of patients with Carney complex. It is a rare feature of McCune-Albright and MEN type I syndrome. With alcohol abuse, the clinical findings of Cushing syndrome may be mimicked, producing the pseudo–Cushing syndrome.
A rapid screening test for Cushing syndrome consists of oral administration of 1 mg of dexamethasone at 11 pm , followed at 8 am by a fluorometric determination of plasma cortisol. A cortisol level below 50 nmol/L essentially rules out Cushing syndrome, except for the iatrogenic variety, in which there is adrenocortical hypoplasia, and the serum cortisol level is very low, even without dexamethasone suppression. If this test is positive, it must be confirmed by doing a 24-hour urinary free cortisol test. A value of at least three times the upper limit of normal is 95%–100% sensitive and specific. A serum ACTH is then obtained to determine whether the source is the adrenal glands or whether it is a pituitary tumor or an ectopic tumor (low, normal or high, and very high, respectively). Treatment is primarily surgical removal of the tumor; however, radiation, chemotherapy, or medication that blocks steroid synthesis is occasionally used.
Al Ojaimi EH: Cushing’s syndrome due to an ACTH-producing primary ovarian carcinoma. Hormones (Athens) 2014; 13: 140.
Brown RJ, et al: Cushing syndrome in the McCune-Albright syndrome. J Clin Endocrinol Metab 2010; 95: 1508.
Ceccato F, Boscaro M: Cushing’s syndrome. High Blood Press Cardiovasc Prev 2016; 23: 209.
Davidovici BB, et al: Cutaneous manifestations of pituitary gland diseases. Clin Dermatol 2008; 26: 288.
Fukuhara D, et al: Iatrogenic Cushing’s syndrome due to topical ocular glucocorticoid treatment. Pediatric 2017; 139: e20161233.
Pichardo-Lowden A, et al: Cushing syndrome related to gingival application of a dexamethasone-containing preparation. Endocr Pract 2010; 16: 336.
Pluta RM, et al: Cushing syndrome and Cushing disease. JAMA 2011; 306: 2742.
Sattar H, et al: Iatrogenic Cushing’s syndrome in children presenting at Children’s Hospital Lahore using nappy rash ointments. J Pak Med Assoc 2015; 65: 463.
Sikorska D, et al: Bilateral primary pigmented nodular adrenal disease as a component of Carney syndrome—case report. Endokrynol Pol 2017; 68: 70.
Addison Disease
Adrenal insufficiency is manifested in the skin primarily by hyperpigmentation ( Fig. 24.3 ). It is diffuse but most prominently observed in sun-exposed areas and sites exposed to recurrent trauma or pressure. The axillae, perineum, and nipples are also affected. Palmar crease darkening in patients of lighter skin type, scar hyperpigmentation, and darkening of nevi, mucous membranes, hair, and nails may all be seen. Multifocal oral melanoacanthoma, a reactive mucosal hyperpigmentation, may occur. An eruptive onset of multiple new nevi may be an early sign of Addison disease. Occasionally, pigmentation may not occur; this is referred to as white Addison disease. Decreased axillary and pubic hair is seen in women, because their androgen production primarily occurs in the adrenals. Fibrosis and calcification of the pinnae of the ears are rare complications.
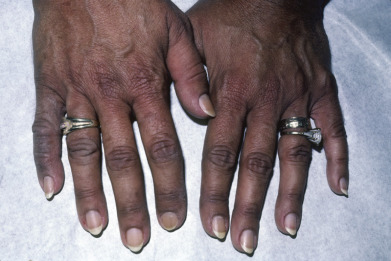
Systemic signs such as weight loss, nausea, vomiting, diarrhea, weakness, fatigue, and hypotension add specificity to the cutaneous abnormalities. Addison disease is usually the result of autoantibody destruction of adrenocortical tissue; however, infection, hemorrhage, or infiltration may be the cause of adrenal insufficiency. In young boys suspected of having Addison disease, adrenoleukodystrophy, a peroxisomal disease, must be considered. Hyperpigmentation associated with adrenocortical insufficiency usually occurs in childhood, and precedes the adult onset of neurologic signs, so very-long-chain fatty acid levels should be determined and analysis for a mutation of the ABCD1 gene done. Addison disease may be part of polyglandular autoimmune syndrome types I, II, and IV, in which various combinations of hypoparathyroidism, chronic candidiasis, vitiligo or autoimmune thyroiditis, and diabetes may occur.
Diagnosis of Addison disease is made by obtaining a paired serum cortisol and plasma ACTH, followed by stimulation with cosyntropin. Failure to see an elevation above 550 nmol/L in 1 hour is diagnostic. Plasma ACTH is elevated in primary insufficiency but normal to low in patients with secondary adrenal insufficiency, in whom the damage is in the hypothalamic-pituitary axis. The adrenals should be imaged with computed tomography (CT) to exclude infiltration or infection.
Treatment of Addison disease is replacement of the glucocorticoids and mineralocorticoids.
Cutolo M: Autoimmune polyendocrine syndromes. Autoimmun Rev 2014; 13: 85.
Dantas TS, et al: Multifocal oral melanoacanthoma associated with Addison’s disease and hyperthyroidism. Arch Endocrinol Metab 2017; 61: 403.
Engelen M, et al: X-linked adrenoleukodystrophy. Curr Neurol Neurosci Rep 2014; 14: 486.
Husebye ES, et al: Consensus statement on the diagnosis, treatment and follow-up of patients with primary adrenal insufficiency. J Intern Med 2014; 275: 104.
Koelemji I, et al: Eruptive melanocytic naevi as a sign of primary adrenocortical insufficiency. Clin Exp Dermatol 2013; 38: 927.
Prat C, et al: Longitudinal melanonychia as the first sign of Addison’s disease. J Am Acad Dermatol 2008; 58: 522.
Combined Pituitary Hormone Deficiency and Growth Hormone Deficiency
Pituitary failure results in many changes in the skin, hair, and nails because of the absence of pituitary hormone action on these sites. Pale, thin, dry skin is seen. Hypohidrosis is present. Diffuse loss of body hair occurs, with axillary, pubic, and head hair being especially thin. The nails are thin, fragile, and opaque and grow slowly. Compromise of the pituitary gland is usually caused by a pituitary tumor, although infiltration, autoimmunity and other inflammatory processes, infection, genetic mutations, trauma, radiation, hemorrhage, or hypothalamic tumors may be the etiology. Isolated GH deficiency is the most common sporadic form of hypopituitarism; however, thyroid hormone, glucocorticoids, and sex steroids may be low and require replacement in combined pituitary hormone deficiency (or panhypopituitarism). A pituitary MRI will screen for tumors or infiltrative processes. Mutations in a large number of genes encoding transcription factors and specific autoantibodies such as anti-pit-1 may also be responsible.
Castinetti F, et al: Combined pituitary hormone deficiency. J Endocrinol Invest 2015; 38: 1.
Davidovici BB, et al: Cutaneous manifestations of pituitary gland diseases. Clin Dermatol 2008; 26: 288.
Di Iorgi N, et al: Classical and non-classical causes of GH deficiency in the paediatric age. Best Pract Res Clin Endocrinol Metab 2016; 30: 705.
Androgen-Dependent Syndromes
The androgen-dependent syndromes are caused by the excessive production of adrenal or gonadal androgens by adrenal adenomas, carcinoma, or hyperplasia, Leydig cell tumors in men, and arrhenoblastomas and polycystic ovarian syndrome (PCOS) in women.
PCOS is defined as the association of polycystic ovaries with biochemical (the best screening laboratory test is free testosterone) or clinical signs of hyperandrogenism and chronic anovulation (less than nine periods per year), without specific underlying disease of the adrenal or pituitary glands. The cutaneous signs of excessive androgen in women include acne, hirsutism, and androgen-induced patterned scalp hair loss. Hyperpigmentation of the skin, areolae, genitalia, palmar creases, and buccal mucosa develops in some patients. Acanthosis nigricans is common in PCOS, reflecting insulin resistance. Women who meet the criteria of PCOS have more severe truncal hirsutism and axillary acanthosis nigricans. Diabetes mellitus, cardiovascular complications, and sleep apnea are associated comorbidities of PCOS. In patients who are obese, weight loss is a prime target of therapy to prevent morbidity and mortality from the associated metabolic syndrome. An association of endometrial cancer is suggested but remains unproven.
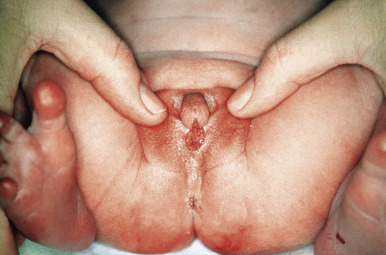
In the congenital adrenogenital syndrome ( Fig. 24.4 ), excess androgen is produced by an inherited defect in any of the five enzymatic steps required to convert cholesterol to cortisol. The formation of inadequate amounts of cortisol stimulates the pituitary to secrete excessive ACTH, which leads to excess androgen production. In boys, precocious puberty results. In girls, masculinization occurs, with the prominent cutaneous signs of excess androgen production. These signs may include acne. Acne with onset between ages 1 and 7 with physical findings suggestive of a hormonal disorder, such as sexual precocity, virilization, and growth abnormalities, should be referred to a pediatric endocrinologist. Acne that begins from ages 7 to 12 often manifests primarily as comedonal lesions in the central face. Unless there are other signs of androgen excess, these patients do not need a workup. Accelerated bone growth with early closure of the epiphyseal plates results in short stature. Early appearance of pubic and axillary hair is also seen.