Cutaneous Lymphoid Hyperplasia (Lymphocytoma Cutis, Lymphadenosis Benigna Cutis, Pseudolymphoma)
Benign cutaneous lymphoid hyperplasia can be caused by medications, injected foreign substances, infections, and arthropod bites, or it may be idiopathic. If there is a histologic resemblance to lymphoma, the term pseudolymphoma is often used. By standard techniques, most cases of cutaneous lymphoid hyperplasia will be found to lack clonality. Cases of monoclonal B-cell and T-cell cutaneous lymphoid hyperplasia do occur. Thus a finding of monoclonality does not equate to the diagnosis of malignancy or lymphoma, and it does not predict biologic behavior.
Two clinical patterns of cutaneous lymphoid hyperplasia exist. The nodular form consists of nodular and diffuse dermal aggregates of lymphocytes, macrophages, and dendritic cells (DCs). The clinicohistologic differential diagnosis is cutaneous B-cell lymphoma. The diffuse type is usually associated with drug exposure or photosensitivity (actinic reticuloid). Histologically, it must be distinguished from cutaneous T-cell lymphoma.
Cutaneous Lymphoid Hyperplasias—Nodular B-Cell Pattern
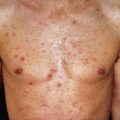
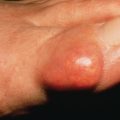
The nodular pattern of cutaneous lymphoid hyperplasia is the most common pattern. It usually presents in adults and is two to three times more common in women. It favors the face (cheek, nose, or earlobe), and the majority of cases present as a solitary ( Fig. 32.1 ) or localized cluster of asymptomatic, erythematous to violaceous papules or nodules. Less frequently, lesions may affect the trunk (36%) or extremities (25%). At times, the lesions may coalesce into a plaque or may be widespread in one region, where they present as miliary papules. Systemic symptoms are absent and, except for rare cases with regional lymphadenopathy, there are no other physical or laboratory abnormalities. It is usually idiopathic but can be caused by tattoos, Borrelia infections, herpes zoster scars, antigen injections, acupuncture, drug reactions, and persistent insect bite reactions.
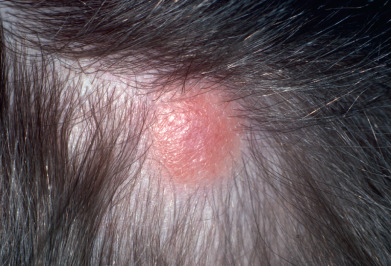
Borrelia -induced cutaneous lymphoid hyperplasia occurs more commonly in young women. It tends to involve the earlobe and nipple. In contrast, borrelial immunocytoma (classified as a subtype of marginal zone lymphoma) appears as tense pink nodules on the legs of older men. The lack of borrelial pseudolymphoma in the United States compared with Europe may relate to the presence of different borrelial species in Europe, specifically Borrelia afzelii, that cause borreliosis. Lesions occur at the site of the tick bite or close to the edge of a lesion of erythema migrans. They may appear up to 10 months after infection. Lesions may be multiple and favor the earlobes, nipple/areola, nose, and, in men, the scrotal area and vary from 1–5 cm in diameter. Usually there are no symptoms, but associated regional lymphadenopathy may be present. Late manifestations of Borrelia infection are uncommon. The diagnosis is suspected from a history of a tick bite or erythema migrans, the location (earlobe or nipple), and the histologic picture. The diagnosis is confirmed by an elevated anti- Borrelia antibody (present in 50% of cases) and the finding of borrelial DNA in the affected tissue. The treatment is penicillin. Rare cases progress to true lymphoma.
Histologic examination of nodular cutaneous lymphoid hyperplasia reveals a dense, nodular infiltrate that occupies primarily the dermis and lessens in the deeper dermis and subcutaneous fat (i.e., it is “top-heavy”). The process is usually separated from the epidermis by a clear grenz zone. The infiltrate is composed chiefly of mature small and large lymphocytes, histiocytes, plasma cells, DCs, and eosinophils. In the deeper portions, well-defined germinal centers are usually seen, with central large lymphoid cells with abundant cytoplasm and tingible body macrophages, and a peripheral cuff of small lymphocytes. A plasma cell–predominant variant has been described. Reactive hyperplasia of adnexal epithelium is common and characteristic, but it may also occasionally be seen in true lymphomas. Germinal centers are symmetric and surrounded by a mix of B and T cells. BCL-6 and CD10 expression is limited to the germinal centers, which also have an intact CD21+ network of DCs. Typically, more than 90% of the cells in the germinal center express the proliferative marker Ki-67 (MIB-1). There is no evidence of light-chain restriction by in situ hybridization. CD30+ cells may occasionally be prominent, raising concern about the development of a CD30+ lymphoproliferative disorder.
Because most lesions are asymptomatic, treatment is often not required. If the process has been induced by a medication, use of the medication should be discontinued. Infection should be treated and localized foci of infection removed. Intralesional steroidal agents are sometimes beneficial, but lesions may recur in a few months. Potent topical corticosteroids may also be tried for superficial lesions. Intralesional corticosteroids, cryosurgery, thalidomide (100 mg/day for a few months), interferon (IFN) alfa, IFN alfa-2b, laser ablation, and surgical excision can all produce good results. Low-dose radiation therapy is usually very effective and may be used on refractory facial lesions that cannot be satisfactorily removed surgically.
Cutaneous Lymphoid Hyperplasias—Bandlike T-Cell Pattern
Cutaneous lymphoid hyperplasias may histologically show a bandlike and perivascular dermal infiltrate, at times with epidermotropism. The lesions may be idiopathic or may be caused by photosensitivity (formerly called actinic reticuloid; now called chronic actinic dermatitis), medications (usually anticonvulsants, but also many others), or contact dermatitis (so-called lymphomatoid contact dermatitis). Clinically these patients have lesions that resemble mycosis fungoides: widespread erythema with scaling. Thicker plaques may occur as well, and these cases are frequently caused by medications. The treatment is to stop any implicated medication. If stopping the medication is ineffective, topical and intralesional corticosteroids, psoralen plus ultraviolet A (PUVA) therapy, and, for persistent localized lesions, radiotherapy may be considered. Histologically, a T-cell–rich band of lymphocytes is present. Epidermotropism, atypia, and even clonality may suggest mycosis fungoides, but the lesions resolve when the drug or other inciting agent is withdrawn.
Jessner Lymphocytic Infiltrate of the Skin
The existence of this entity has recently been challenged, and the condition may best be classified as a variant of lupus erythematosus (LE). Clinically Jessner infiltrate is a persistent papular and plaquelike eruption that is photosensitive and occurs primarily on the face. Histologically there is a superficial and deep perivascular and periadnexal lymphocytic infiltrate. Interface dermatitis is absent. The infiltrating lymphocytes are suppressor T cells (CD8+). Features that suggest this may be distinct from other forms of cutaneous LE include the absence of an interface dermatitis, lack of mucin, and negative direct immunofluorescence (DIF). Tumid LE also lacks interface dermatitis but has ample mucin. Polymorphous light eruption (PMLE) is distinguished from Jessner infiltrate by having edematous papules and plaques that are more transient and by the presence of dermal edema. In PMLE the infiltrating cells are also CD8+. True cases of lymphocytic infiltration of the skin may still exist. To distinguish them clearly from LE and PMLE, the lesions must contain predominantly CD8+ suppressor T cells, must lack dermal mucin and dermal edema, and must be fixed (not transient as with PMLE); patients must have negative DIF and serologic testing for LE. Both Jessner lymphocytic infiltrate and chronic cutaneous LE respond to antimalarials.
Mitteldorf C, et al: Cutaneous pseudolymphoma. Surg Pathol Clin. 2017; 10: 455.
Romero-Pérez D, et al: Cutaneous pseudolymphomas. Actas Dermosifiliogr 2016; 107: 640.
Shetty SK, et al: Pseudolymphoma versus lymphoma. J Oral Maxillofac Pathol 2016; 20: 328.
Staser K, et al: Injection-site cutaneous pseudolymphoma induced by a GM-CSF-producing tumor cell vaccine. JAMA Dermatol 2017; 153: 332.
Cutaneous Lymphomas
Because cutaneous Hodgkin disease is very rare, the term non-Hodgkin lymphoma has little meaning when speaking of a lymphoma in the skin, because virtually all cutaneous lymphomas are “non-Hodgkin lymphomas.” Cutaneous lymphoma can be considered to be either primary or secondary. Primary cutaneous lymphomas are those that occur in the skin, and where no evidence of extracutaneous involvement is found for some period after the appearance of the cutaneous disease. Secondary cutaneous lymphoma includes cases that have simultaneous or preceding evidence of extracutaneous involvement. These cases are best classified and managed as lymph node–based lymphomas with skin involvement. This conceptual separation is not ideal, but it has been important in developing classification schemes and determining prognosis in cutaneous lymphomas.
For many years, classification of lymphomas has been based on their histologic appearance, and lesions from all organ systems were classified histomorphologically in an identical manner to lymphomas arising in lymph nodes. It had been recognized that these classification schemes have major shortcomings when applied to extranodal lymphomas. Specifically, they did not uniformly predict clinical behavior. The new World Health Organization (WHO) classification scheme recognizes distinct forms of primary cutaneous lymphoma.
Cutaneous lymphomas are classified based on their cell type. There are B-cell lymphomas and T-cell lymphomas, but B-cell lymphomas can be T-cell rich. In the latter cases, atypia is restricted to the B-cell population, and immunoglobulin gene rearrangements are detected. Histologic features used in the classification system include cell size (large vs. small), nuclear morphology (cleaved or noncleaved), and immunophenotype. Because appropriate classification may be prognostically important, experienced dermatopathology consultation should be sought in cases of cutaneous lymphoma.
Primary Cutaneous T-Cell Lymphomas
A major insight into cutaneous lymphoma was the finding that the majority of lymphomas in the skin were of T-cell origin. This is logical, because T cells normally traffic through the skin and are important in “skin-associated lymphoid tissue.” Unfortunately, dermatologists frequently use the term cutaneous T-cell lymphoma (CTCL) synonymously with mycosis fungoides (MF). Although MF represents the large majority of primary CTCLs, up to 30% of primary CTCLs are not MF. The following discussion is divided into MF and related conditions, Sézary syndrome, lymphomatoid papulosis, and non-MF primary CTCLs.
Mycosis Fungoides
Mycosis fungoides (MF) is a malignant neoplasm of T-lymphocyte origin, almost always a memory T-helper (Th) cell. The incidence has been cited as 1 in 300,000 per year, but has been increasing. MF affects all races. In the United States black persons are relatively more often affected than white persons. MF is twice as common in men as in women.
Natural History.
In most cases, MF is a chronic, slowly progressive disorder. It usually begins as flat patches (patch stage) favoring the bathing suit area and lower trunk. Biopsy may not be diagnostic at this stage, but the inability to diagnose early cases has more to do with the limits of diagnostic capabilities than a transformation from some nonneoplastic (premycotic) condition to MF, and these cases are best considered MF from the onset. Pruritus, sometimes severe, is usually present at this stage. Over time, sometimes years, the lesions become more infiltrated, and the diagnosis is usually confirmed with repeated histologic evaluation. Infiltrated plaques occur eventually (plaque stage). In some cases, tumors may eventually appear (tumor stage). Some MF patients may present with or progress to erythroderma. Most rarely, patients may present with tumors de novo, the so-called d’emblée form. With immunophenotyping, many MF cases are now recognized as non-MF T-cell lymphomas. Eventually, in some patients noncutaneous involvement is detected. This is usually first identified in lymph nodes. Peripheral blood involvement and visceral organ involvement may also occur.
In general, MF affects elderly patients and has a long evolution. However, once tumors develop or lymph node involvement occurs, the prognosis is guarded, and MF can be fatal. In most fatal cases, the patient dies of septicemia. Early, aggressive chemotherapy in an attempt to “cure” MF is associated with excessive morbidity and mortality rates and is not indicated.
Evaluation and Staging.
Staging information can be found at http://www.cancer.gov/types/lymphoma/hp/mycosis-fungoides-treatment-pdq . Because MF is a systemic disease from the onset (because lymphocytes naturally traffic throughout the body), concepts used for solid tumors, such as tumor burden and metastasis, cannot be readily applied. The TNMB system scores involvement in the skin (T), lymph node (N), viscera (M), and peripheral blood (B) and is in evolution. Skin involvement is divided into less than 10% (T1), more than 10% (T2), tumors (T3), and erythroderma (T4). Node involvement is normal clinically and pathologically (N0), palpable but pathologically not MF (N1), not palpable but pathologically MF (N2), or clinically and pathologically involved (N3). Viscera and blood are either not involved (M0 and B0) or involved (M1 and B1).
- •
Stage IA is T1, N0, M0.
- •
Stage IB is T2, N0, M0.
- •
Stage IIA is T1–T2, N1, M0.
- •
Stage IIB is T3, N0–N1, M0.
- •
Stage IIIA is T4, N0, M0.
- •
Stage IIIB is T4, N1, M0.
- •
Stage IVA is T1–T4, N2–N3, M0.
- •
Stage IVB is T1–T4, N0–N3, M1.
The “B” or blood status does not alter staging of the disease. The International Society for Lymphomas/European Organization of Research and Treatment of Cancer staging has differed by putting all clinically normal nodes into N0 and indicating the presence or absence of a clone in node or blood.
Example:
N1a: Clone negative
N1b: Clone positive
A staging workup would include a complete history and physical examination, with careful palpation of lymph nodes and mapping of skin lesions; a complete blood cell count (CBC) with assays for circulating atypical cells (Sézary cells); serum chemistries, including renal and liver function tests with lactate dehydrogenase; a chest radiograph evaluation; and a skin biopsy. If palpable, lymph nodes should be examined histologically. Fine-needle aspiration is not an ideal mode of evaluation, because early lymph node involvement may be localized to certain areas of the affected nodes and often requires architectural evaluation for detection. If any abnormalities are detected through these evaluations, they should be pursued. Computed tomography (CT) can be performed to assess chest, abdominal, and pelvic lymph nodes and visceral organs. These tests are useful in patients with stage II to IV disease, but are not indicated in patients with stage IA disease. Whether patients with stage IB disease should undergo these tests is unknown.
Stage IA patients have a life expectancy identical to that of a control population; only 8%–9% progress to more advanced disease, and only 2% die of their disease. By contrast, patients with T2 disease have shorter survival than controls (median survival of 11.7–15.6 years); 24% of T2 patients progress to more advanced disease. T3 patients have a median survival of 3.2–8.4 years, and T4 patients, 1.8–3.7 years. Palpable adenopathy is associated with a median survival of only 7.7 years, whereas patients without adenopathy have survival of 21.8 years. Lymphadenopathy, tumors, and cutaneous ulceration are cardinal prognostic factors; no patient dies without having developed one of these, and patients with all three (in any order) survive a median of 1 year.
Clinical Features.
In the early patch/plaque stage, the lesions are macular or slightly infiltrated patches or plaques varying in size, with many measuring 5 cm or more. Folliculotropic disease can resemble lichen nitidus or lichen spinulosis. Except for the folliculotropic and childhood variants, lesions greater than 5 cm are virtually always present. In contrast, most histologic simulators present with smaller skin lesions. The eruption may be generalized or may begin localized to one area and then spread. The lower abdomen, buttocks, upper thighs, and breasts of women are preferentially affected. The lesions may have an atrophic surface or may present as true poikiloderma with atrophy, mottled dyspigmentation, and telangiectasia. Poikiloderma vasculare atrophicans most often represents a clinical form of patch-stage MF. Likewise, large-plaque parapsoriasis and cases of small-plaque parapsoriasis with poikilodermatous change are early patch-stage lesions of MF. In contrast, typical digitate dermatosis rarely if ever evolves into MF. “Invisible” MF is generalized skin involvement that is not visible to the naked eye but can be documented histologically. With current diagnostic methods, this can usually be confirmed. In general, the patch-stage lesions ( Fig. 32.2 ) resemble eczema. Ovoid, annular, polycyclic, or arciform configurations can occur. Less common forms are the verrucous or hyperkeratotic form, the hypopigmented form (which predominates in dark-skinned children) ( Fig. 32.3 ), lesions resembling a pigmented purpura, and the vesicular, bullous, or pustular form. Subtle lesions of MF may manifest clinically during anti–tumor necrosis factor (TNF) therapy.
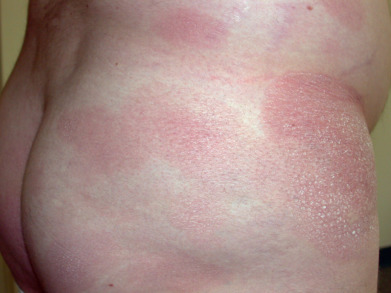
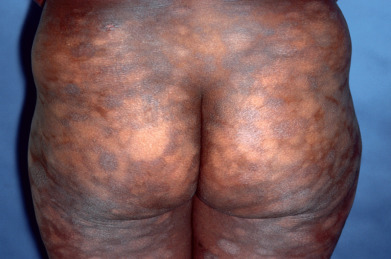
In the plaque stage lesions are more infiltrated and may resemble psoriasis ( Fig. 32.4 ), a subacute dermatitis, or a granulomatous dermal process such as granuloma annulare. The palms and soles may be involved, with hyperkeratotic, psoriasiform, and fissuring plaques. The infiltration of the plaques, at first recognized by light palpation, may be present in only a few of the lesions. It is a manifestation of diagnostic importance. Different degrees of infiltration may exist, even in the same patch, and sometimes it is more pronounced peripherally, the central part of the plaque being depressed to the level of the surrounding skin. The infiltration becomes more marked and leads to discoid patches or extensive plaques, which may be as wide as 30 cm.
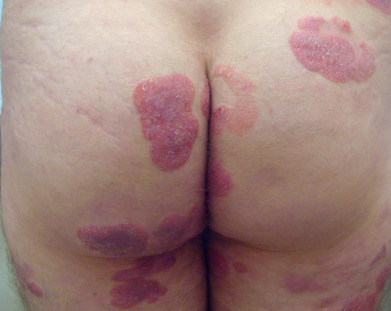
Eventually, through coalescence of the various plaques, the involvement becomes widespread, but there are usually patches of apparently normal skin interspersed. When the involvement is advanced, painful superficial ulcerations may occur. During this phase, enlarged lymph nodes usually develop. They are nontender, firm, and freely movable.
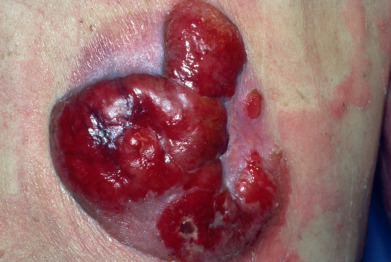
The tumor stage is characterized by large, variously sized and shaped nodules on infiltrated plaques ( Fig. 32.5 ) and on apparently normal skin. These nodules tend to break down early and to form deep oval ulcers, whose bases are covered with a necrotic grayish substance and which have rolled edges. The lesions generally have a predilection for the trunk, although they may be seen anywhere on the skin or may involve the mouth and upper respiratory tract. Infrequently, tumors may be the first sign of MF.
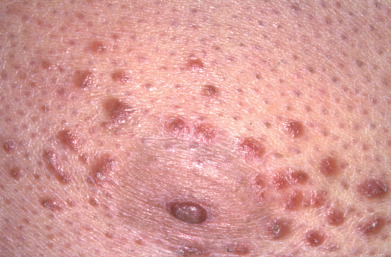
The erythrodermic variety of MF is a generalized exfoliative process that often exhibits islands of spared skin. The hair is scanty, nails are dystrophic, palms and soles are hyperkeratotic, and at times, generalized hyperpigmentation may occur. Erythroderma may be the presenting feature.
Alopecia Mucinosa.
The infiltrating cells of MF can demonstrate a predilection for involving the hair follicle ( Fig. 32.6 ). This may be observed simply by folliculotropism of the cells (pilotropic or follicular MF) or by the appearance of follicular mucinosis ( Fig. 32.7 ). In all cases of follicular mucinosis, the histologic specimen should be carefully examined and the diagnosis of MF considered. Among patients older than 40 who have follicular mucinosis, a large percentage will have MF or go on to develop it. However, the finding of a T-cell clone in lesions of follicular mucinosis without MF is not predictive of the development of CTCL.
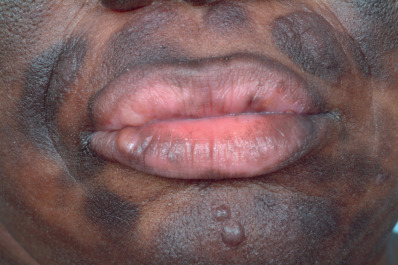
Selective tropism of the CTCL cells to the sweat glands and ducts is termed syringotropic CTCL ( Fig. 32.8 ). This is often seen in conjunction with follicular involvement. Syringolymphoid hyperplasia may be seen in these cases histologically and may mimic eccrine carcinoma. Cases previously called “syringolymphoid hyperplasia with alopecia” are now considered to be cutaneous T-cell lymphoma. Clinically, the lesions present as discrete follicular and nonfollicular erythema, along with alopecia, milia, and follicular cysts. The initial clinical diagnosis in such cases is often discoid lupus erythematosus. The prognosis in MF with adnexal involvement is as predicted by the staging system for other MF forms. Patients with granulomatous MF have a poorer prognosis and a poorer response to skin-directed therapy.
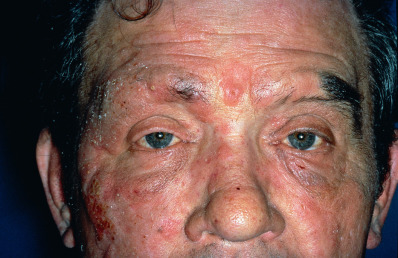
Systemic Manifestations.
MF as a form of malignant lymphoma may progress to include visceral involvement. Lymph node involvement is most common; it predicts progression of MF in at least one quarter of patients and reduces survival to about 7 years. Any other evidence of visceral involvement is a poor prognostic sign. An abnormal result on liver-spleen scan, chest radiograph or CT evaluation, abdominal or pelvic CT scans, or bone marrow biopsy is associated with a survival of about 1 year. The prognosis is worse in non-Caucasian patients with early-onset MF, especially African American women.
Pathogenesis.
MF is a neoplasm of memory Th cells in most cases. Rare cases of suppressor cell (CD8+) MF have been reported. These CD8+ cases may behave indolently, like MF, or aggressively. The aggressive subset tends to present with plaques rather than patches. The events leading to the development of the malignant T cells are unknown. Some speculate that it is caused by chronic exposure to an antigen, but this has yet to be confirmed. Patients with atopic dermatitis appear to be at increased risk for development of MF, suggesting that persistent stimulation of T cells may lead to development of a malignant clone.
The inflammatory nature of the skin lesions has led to investigation of the interactions of the malignant T cells and both keratinocytes and antigen-presenting cells (APCs, including Langerhans cells) in MF. MF skin lesions have many features of skin that is immunologically “activated.” MF cells express cutaneous lymphocyte antigen (CLA), the ligand for E selectin, which is expressed on the endothelial cells of inflamed skin. This allows the malignant cells to traffic into the skin from the peripheral blood. CCR4, another homing molecule, is expressed on MF cells, and the ligand for this receptor is on basal keratinocytes. APCs are increased in MF lesions and have increased functional capacity to activate T cells. There is increased expression of major histocompatibility complex (MHC) class II antigens on the surface of the APCs. Through cytokines, infiltration of neoplastic and reactive T cells is increased. The pattern in early MF is more Th1-like, and the nonneoplastic infiltrating cells (tumor-infiltrating lymphocytes, TILs) may play a role in downregulating and controlling the neoplastic cells. There are more CD8+ cells in these early lesions, and these TILs may control the malignant clone. In fact, MF patients with more than 20% CD8+ cells in their skin survive longer than those with less than 15%. In more advanced MF and in Sézary syndrome, perhaps through interleukin (IL)–4 and IL-10, a Th2 environment exists. This downregulates suppressor cell function and allows the malignant clone to proliferate. In addition, the Th2-dominant environment reduces effective Th-cell function, explaining the increased risk of infection and secondary cancer in patients with advanced CTCL. Correcting the aberrant immune response in advanced CTCL is the basis of some treatment approaches.
Common chromosomal alterations in MF include gain of 7q36 and 7q21–7q22 and loss of 5q13 and 9p21. This characteristic pattern differs from that seen in Sézary syndrome, suggesting that the two disorders are distinct. As MF advances, the number of circulating malignant T cells increases, and they can be detected by flow cytometry as CD4+,CD7− or CD4+,CD26− circulating cells.
Histopathology.
Perhaps more than in any other situation in dermatopathology, the ability to diagnose MF histologically correlates closely with the skill, training, and experience of the reviewing pathologist. When the clinician is considering a diagnosis of MF, consultation with a skilled dermatopathologist should be strongly considered if original histologic reports are nonconfirmatory or nonspecific.
In patch-stage lesions, subtle epidermotropism of lymphocytes resembles a vacuolar interface dermatitis with a lymphocyte in every vacuole. As lesions progress, there is a distinct bandlike distribution of lymphocytes with epidermotropism. At this stage, a large, dark lymphocyte is present in every vacuole. The lymphocytes within the epidermis may be numerous or few but are typically larger, darker, and more angulated than those in the dermis. Papillary dermal fibrosis is typically present. The superficial perivascular lymphoid infiltrate that surrounds the postcapillary venule is typically more prominent above the vessel than below the vessel (“bare underbelly” sign).
Plaques of MF show a more prominent, superficial bandlike lymphoid infiltrate and a deeper perivascular dermal component than patch-stage lesions. Papillary dermal fibrosis is more prominent, and the subpapillary plexus is shifted downward. Epidermotropism is much more marked and is typically associated with minimal spongiosis. This helps distinguish patch-stage MF from spongiotic dermatitis. Vesicular variants are an exception to this rule. In vesicular variants, spongiosis is prominent and results in intraepidermal and subcorneal vesiculation. Eosinophils are common in folliculotropic MF (with or without follicular mucinosis) but are uncommon in other MF forms.
In thick plaques and tumors, epidermotropism may be substantially diminished. The diagnosis of MF is confirmed by the presence of dense sheets of infiltrating lymphocytes in the dermis and subcutaneous fat. These cells may have cerebriform nuclei.
Cardinal features that should suggest a diagnosis of MF include the following:
- •
Solitary or small groups of lymphocytes in the basal cell layer
- •
Epidermotropism of lymphocytes, with disproportionately scant spongiosis
- •
More lymphocytes within the epidermis than would normally be seen in an inflammatory dermatosis, with little accompanying acanthosis or spongiosis
- •
Lymphocytes in the epidermis larger than those in the dermis
- •
Papillary dermal fibrosis with bundles of collagen arranged haphazardly
- •
Prominent folliculotropism or syringotropism of the lymphocytes, especially with intrafollicular mucin deposition (follicular mucinosis)
Features that should suggest a diagnosis of inflammatory dermatosis over MF include the following:
- •
Prominent upper dermal and papillary edema
- •
Marked epidermal spongiosis
- •
Accumulation of the intraepidermal inflammatory cells in flask-shaped collections, with the top open to the stratum corneum
Immunohistochemistry is of some value in assessing MF. MF cells characteristically are CD4+, but lose the CD7 and CD26 antigens. Loss of CD7 expression within the large, dark lymphocytes in the epidermis, with normal expression in the benign recruited lymphocytes in the infiltrate below, suggests a diagnosis of MF. DNA hybridization or a Southern blot test is frequently performed in equivocal cases to detect clonal rearrangement of the T-cell receptor (TCR). An identical rearrangement at multiple sites suggests MF. In early lesions of MF, the number of infiltrating cells may be insufficient for a clone to be detected, so a negative test does not exclude the diagnosis of MF. Testing with fresh tissue is somewhat more sensitive than with fixed tissue using current methods. Similar techniques can be used to evaluate lymph nodes in MF patients. Lymph node involvement can be detected by these molecular methods, whereas routine histologic evaluation yields normal results. Patients with more advanced disease are more likely to have clones in their lymph nodes, and the presence of clonality is predictive of shorter survival.
Differential Diagnosis.
In the early patch stage, MF may be difficult to diagnose. The skin lesions usually resemble a nondescript form of eczema with some scale. Interestingly, despite the itching, scratch marks and lichenification are usually absent. MF presenting as papuloerythroderma of Ofuji is an obvious exception. The multiple morphologies of MF make the differential diagnosis vast. Plaquelike lesions may resemble subacute dermatitis or psoriasis. Tumors must be differentiated from other forms of lymphoreticular malignancy and metastases.
Treatment.
Many forms of therapy induce remissions of variable length. The therapeutic choice depends on extent of disease, the patient’s overall health and physical status, the physician’s experience and preference, and the availability of various options. The new topical biologic response modifier resiquimod has shown dramatic effects in many patients and may become first-line therapy. Other agents are still commonly used, as the disease presents with a wide spectrum. Topical corticosteroids, topical nitrogen mustard or 1,3-bis-(2-chloroethyl)-l-nitrosourea (carmustine, BCNU), bexarotene gel 1%, and PUVA or narrow-band UVB are generally good choices for stages IA, IB, and IIA disease. Patch-stage MF has responded to alefacept. Total-skin electron beam (TSEB) therapy can be used for refractory stage IIA and IIB cases. Single-agent chemotherapy or photophoresis can be used as initial management for stage III patients. Low-dose methotrexate may control the skin lesions of MF but has been associated with development of a secondary aggressive lymphoma in a few patients. Pegylated liposomal doxorubicin and combinations of IFN alfa, retinoids (bexarotene or isotretinoin), photophoresis, IFN gamma, skin-directed PUVA, sargramostim (granulocyte-macrophage colony-stimulating factor), alemtuzumab, and perhaps IL-2, IL-12, and IFN alfa may be effective in stage IV disease, as well as for patients who have failed the therapies previously cited for stages IIB and III MF. Multiagent systemic chemotherapy is used much less often with the advent of immunomodulatory treatments for MF. Chemotherapy should be considered only when all other treatment options have failed. Treatment of early-stage disease is in general restricted to skin-directed treatments. More advanced disease is treated with different modalities at different institutions. Combinations of agents are often used, and the combinations and their order of use vary among institutions. In general, therapies that also enhance the patient’s immune system are favored in persons with more advanced disease. Complete remission of MF has been noted after a severe reaction to combined therapy with bexarotene, vorinostat, and high-dose fenofibrate. The reaction included fever, extensive skin necrosis, and granuloma formation.
Biologic Response Modifiers (Multimodality Immunomodulatory Therapy).
Resiquimod and imiquimod may completely change the treatment of CTCL, as therapy directed at a few lesions can produce widespread regression of the disease. IFN alfa and IFN gamma have been shown to have efficacy against MF. IFN alfa is associated with a positive response in about 60% of patients and a complete response in 19%. Toxicity is significant and includes fever, chills, myalgias, neutropenia, and depression. Low-dose IFN-alfa and IFN-gamma treatments and granulocyte-macrophage colony-stimulating factor (GM-CSF) are now used in adjunctive fashion in combination with retinoid therapy, phototherapy, and other modalities. This is termed multimodality immunomodulatory therapy.
Topical Corticosteroids.
The availability of superpotent class I topical corticosteroids has led to a reassessment of their possible role in the management of early MF (patch stage, T1 and T2). Zackheim et al. reported a 63% complete remission rate for patients with T1 disease and a total response rate of 94%. In T2 patients, complete response was seen in only 25% but total response in 82%. The predominant side effect was a temporary and reversible suppression of the hypothalamic-pituitary axis in about 13% of patients.
Topical Nitrogen Mustard.
Anhydrous gel or ointment-based mustard products are being used more often, but aqueous mustard is still used as well, with a 10-mg vial of mechlorethamine hydrochloride dissolved in 60 mL of tap water and applied to the entire skin surface, except the face, axillae, and genitalia, with a 2-inch paint brush or gauze pad. The last milliliter may be diluted to half-strength or greater dilution for application to the face, axillae, and genitalia. Such treatment leads to complete responses in 80% of patients with stage IA disease, 68% in stage IB, 61% in stage IIA, 49% in stage IIB, and 60% in stage III patients. About 10% of patients obtain a durable and long-lasting remission of more than 8 years. The major side effects of topical nitrogen mustard (NH2) therapy are cutaneous intolerance, which occurs in almost 50% of patients, and allergic contact dermatitis, which occurs in 15%. Short (1 hour) contact does not reduce this rate of sensitization. This can be reduced by the use of an ointment formulation, but response rates have been reported to be inferior with the ointment form. At least half of patients will relapse when therapy is stopped, but they frequently will respond again to NH2.
The duration of maintenance therapy after achieving remission varies in different centers. Some treat for an additional 6 months, and others taper treatment over 1 year or more, or continue treatment indefinitely.
Topical BCNU (Carmustine).
Topical BCNU, 2 mg/mL in 150-mL aliquots, dissolved in ethanol, is dispensed to the patient. From this stock solution, the patient takes 5 mL and adds it to 60 mL of water at room temperature. This is applied once a day to the whole body, sparing the folds, genitals, hands, and feet (if they do not have lesions). If the extent of disease is limited, only the affected areas are treated. The average treatment course is 8–12 weeks. If, after 3–6 months, the patient’s condition is not responding, the concentration may be doubled and the treatment repeated for 12 weeks. For small or persistent lesions, the straight stock solution may be applied daily. Patients tolerate BCNU better than nitrogen mustard, contact sensitization is uncommon, and responses are more rapid. CBC should be monitored monthly during treatment, but marrow suppression occurs in less than 10% of patients treated with the low concentrations. Telangiectasia, which may be persistent and severe, can occur after prolonged BCNU therapy or after an adverse cutaneous reaction to the medication.
Ultraviolet Therapy.
Both UVB (narrow- or broad-band) and PUVA (systemic or bath) have been effective in the management of MF. About 75% of patients with patch-stage disease will have a complete clinical remission with UVB therapy. Home therapy is successful. PUVA has been used more extensively and, because of its deeper penetration, is perhaps better suited to the treatment of a disorder with a dermal component. Complete clearing is seen in 88% of patients with limited patch/plaque disease and in 52% of patients with extensive disease. Tumor-stage MF patients do not typically clear, and erythrodermic patients have poor tolerance for PUVA. Up to 50% of patients with a complete response to PUVA may have a remission of up to 10 years. Retinoids and IFN alfa may be added to PUVA. Retinoids may reduce the total number of PUVA treatments required. Low-dose IFN alfa plus PUVA may be used in patch-stage patients in whom topical therapy and PUVA alone are ineffective. The excimer laser may be used once or twice a week to deliver the phototherapy if the patient has a limited number of lesions. On average, 5–6 weeks of treatment is required, and remissions of up to 2 years or more can be achieved.
Extracorporeal photochemotherapy (photophoresis, ECP) is a therapeutic modality in which the circulating cells are extracted and treated with UVA outside the body; the patient ingests psoralen before the treatment. Complete responses are seen in about 20% of MF patients, and a partial response occurs in a similar percentage. In the original reports, the overall response rate for erythrodermic patients was 80%, but many of these patients failed to have at least the 50% clearing required to be considered a partial response. In one comparative trial, standard PUVA was significantly more effective than photophoresis alone, and photophoresis was judged ineffective in plaque-stage (T2) MF. ECP is now used in combination with other agents, especially IFN alfa, and appears to have better efficacy. Insulin-dependent diabetic patients respond poorly.
Photodynamic Therapy.
Photodynamic therapy (PDT) with methyl-aminolevulinic acid has been used successfully for paucilesional MF. Responses were seen in 75% of patients, and patient satisfaction was high. Other photosensitizing agents are being evaluated.
Radiation.
TSEB therapy in doses in excess of 3000 Gy is very effective in the management of MF. Stage T1 patients have a 98% complete response; stage T2, 71%; stage T3, 36%; and stage T4, 64%. Long-term remissions occur in about 50% of T1 patients and 20% of T2 patients. Erythrodermic patients tolerate TSEB therapy poorly; other modalities should be attempted initially. Adjuvant therapy with a topical agent or PUVA can be considered if the patient relapses, as frequently occurs. The most common side effects of TSEB therapy are erythema, edema, worsening of lesions, alopecia, and nail loss. Persistent hyperpigmentation and chronically dry skin are also problems after TSEB therapy. Orthovoltage radiation may be used to control tumors or resistant thick plaques in patients whose conditions have been otherwise controlled with another modality.
Retinoids.
Both isotretinoin and etretinate have efficacy in the treatment of MF. A clinical response is noted in about 44% of patients. Dosage of isotretinoin is about 1 mg/kg/day to start and may be increased up to 3 mg/kg/day as tolerated. Retinoids may be effective in stage IB (T2) and stage III patients, and as a palliative treatment in those with stage IVA disease. Bexarotene (Targretin), a synthetic retinoid that is bound preferentially by the retinoid X receptor (RXR), is thought to work by inducing apoptosis in the malignant T cells. It is available as a topical gel and as an oral tablet. Topical therapy is used in patients with stage IA to IIA CTCL. Patients improve about 50% with this treatment. Oral bexarotene at a dose of 300 mg/m 2 also has a response rate of about 50% in early-stage CTCL. This dose is complicated by hypercholesterolemia, marked hypertriglyceridemia (at times complicated by pancreatitis), central hypothyroidism, and leukopenia. It may be combined with PUVA and other forms of treatment at a lower dose.
Systemic Chemotherapy.
Multidrug chemotherapy often exacerbates the ongoing immune imbalance and may prevent the patient’s immune system from attacking the malignant T cells. For this reason, and because of the enhanced efficacy of combination immunomodulatory treatment regimens, systemic chemotherapy is now very uncommonly used for MF. Methotrexate alone, in doses from 5–125 mg/wk, is effective for the management of T3 patients. In these patients Zackheim et al. reported that 41% had a complete response, and an additional 17% a partial response, giving a total response of 58%. The median overall survival was 8.4 years, and 69% of patients were alive at 5 years. For advanced MF, higher doses of methotrexate with citrovorum-factor rescue were successful in obtaining a response, which was then maintained with lower doses of methotrexate, not requiring rescue. Similarly, vorinostat (and other histone deacetylase inhibitors), pentostatin, etoposide, fludarabine, and 2-chlorodeoxyadenosine have been used. Curcumin is being studied, as are forodesine, a novel inhibitor of purine nucleoside phosphorylase, and pralatrexate, a novel targeted antifolate agent.
Fusion Toxin.
The toxin DAB389IL-2 is the fusion of a portion of the diphtheria toxin to recombinant IL-2. This selectively binds to cells expressing the IL-2 receptor and leads to their death. A series of MF patients who expressed the IL-2 receptor demonstrated a response rate of 37%, including a complete response in 14%. These patients had failed conventional therapies. Patients in stages I to III achieved response, but no patient with stage IV disease did so. Fever, chills, hypotension, nausea, and vomiting were common, and at high doses a vascular leak syndrome occurred. This agent is reserved for advanced-stage patients who have failed other modalities.
Ahn CS, et al: Mycosis fungoides. Am J Dermatopathol 2014; 36: 933.
Berg S, et al: Multidisciplinary management of mycosis fungoides/Sézary syndrome. Curr Hematol Malig Rep 2017; 12: 234.
Boulos S, et al: Clinical presentation, immunopathology, and treatment of juvenile-onset mycosis fungoides. J Am Acad Dermatol 2014; 71: 1117.
Hoppe RT: Remarkable advances in the management of mycosis fungoides and the Sezary syndrome. Oncol Res Treat 2017; 40: 242.
Kelati A, et al: Defining the mimics and clinico-histological diagnosis criteria for mycosis fungoides to minimize misdiagnosis. Int J Womens Dermatol 2017; 3: 100.
Lewis DJ, et al: Complete resolution of mycosis fungoides tumors with imiquimod 5% cream. J Dermatolog Treat 2017; 28: 567.
Vaidya T, et al: Mycosis Fungoides. 2018 Oct 1. StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2018 Jan-. Available from http://www-ncbi-nlm-nih-gov.easyaccess2.lib.cuhk.edu.hk/books/NBK519572/PubMed PMID: 30137856 .
Vonderheid EC, et al: CD4(+)CD26(–) lymphocytes are useful to assess blood involvement and define B ratings in cutaneous T cell lymphoma. Leuk Lymphoma 2018; 59: 330.
Pagetoid Reticulosis
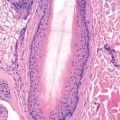
Localized epidermotropic reticulosis, pagetoid reticulosis, or Woringer-Kolopp disease is an uncommon lymphoproliferative disorder considered be a form of mycosis fungoides. Other terms suggested for these cases have included acral mycosis fungoides or mycosis fungoides palmaris et plantaris. In large, MF clinics, such cases represent about 0.6% of all MF cases. Pagetoid reticulosis is divided into classic Woringer-Kolopp, which usually describes solitary lesions, and cases with multiple lesions (Ketron-Goodman variant). The unique features of Woringer-Kolopp disease are clinical. The disease presents as a solitary lesion that is often located on an extremity and frequently has a keratotic rim. If there is more than a single lesion, the lesions often tend to involve both the palms and the soles. Frequently, over months to years, the lesion gradually enlarges, reaching more than 10 cm in size. In some cases, the lesions spontaneously come and go over many years. About 20% of cases occur in patients who are younger than 15 years. The long duration without progression has been a clinical hallmark of Woringer-Kolopp disease. Histologically, there is prominent epidermotropism of lymphocytes, with many lining up in the basal cell layer. This histologic pattern correlates with strong αEβ7- and α4β7-integrin expression by the infiltrating cells. This integrin expression is also seen in the epidermotropic cells of classic MF and contact dermatitis. In MF, most cases are CD4+, but in the acral MF cases, they may be CD4+, CD8+, or negative for both. TCR gene rearrangements can be detected in many cases of Woringer-Kolopp disease. Therapeutically, local excision and radiation therapy have been “curative” in many patients. Topical and systemic PUVA and PDT have also proved effective. Local recurrence is possible.
Sézary Syndrome
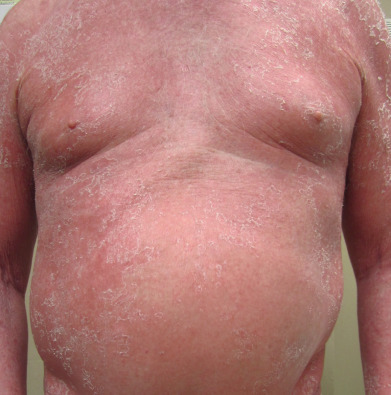
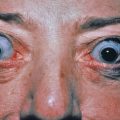
Sézary syndrome is the leukemic phase of mycosis fungoides. The characteristic features are generalized erythroderma ( Fig. 32.9 ), superficial lymphadenopathy, and atypical cells in the circulating blood. Although patients with classic MF may progress to Sézary syndrome, patients with Sézary syndrome usually are erythrodermic from the onset. The skin shows a generalized or limited erythroderma of a typical fiery red color. Associated features can include leonine facies, eyelid edema, ectropion, diffuse alopecia, hyperkeratosis of the palms and soles, and dystrophic nails. Some patients develop lesions identical to vitiligo, especially on the lower legs. Symptoms include severe pruritus and burning, with episodes of chills. Prognosis is poor, with an average survival of about 5 years.