Abstract
Craniosynostosis refers to the premature fusion of one or more cranial sutures. This often results in characteristic cranial distortions in infancy that prompt investigation with imaging. Once craniosynostosis is confirmed, surgical intervention may include craniectomy, frontoorbital advancement, posterior cranial vault remodeling, total cranial vault remodeling, or posterior vault distraction. The intervention is chosen based on the sutures involved, severity of symptoms, and if an underlying syndrome is present. A discussion of postoperative care guidelines and outcomes conclude the chapter.
14 Craniosynostosis
14.1 Goals and Objectives
Recognize the key historical and physical findings needed for diagnosing and treating craniosynostosis.
Clearly define the objectives of surgical treatment of craniosynostosis.
Appreciate the complex surgical procedures utilized in treating patients with craniosynostosis, including the appropriate indications and timing of these procedures.
Understand the evidence-based preoperative and postoperative concerns related to craniosynostosis.
14.2 Patient Presentation
During the first year of life, infants are frequently referred to a plastic surgeon or neurosurgeon for an evaluation of their head shape. The primary concern of the referring provider is usually underlying craniosynostosis, or the premature fusion of one or more cranial sutures. Craniosynostosis may occur as the result of an isolated, sporadic genetic mutation or as part of a complex syndromic diagnosis. Overall, craniosynostosis occurs in 1:2,000, and syndromic craniosynostosis is even more rare, affecting 1:25,000 to 1:100,000 infants. 1 The most common type of craniosynostosis is sagittal synostosis, followed by metopic synostosis, then unicoronal synostosis. 1 Since the “Back to Sleep” campaign began by the American Academy of Pediatrics in 1992, deformational plagiocephaly is much more common than craniosynostosis, nearly as frequent as 1:12. 1 , 2 , 3 Positional plagiocephaly is managed nonsurgically with observation or helmeting, while craniosynostosis is often treated surgically. This makes correctly diagnosing craniosynostosis of critical importance.
History and physical examination will assist in determining if further diagnostic workup is necessary. Neonatal and gestational history including pregnancy complications and exposures should be obtained. A description of the child’s head shape at birth and subsequent shape changes leading to the present visit is important in assessing if the deformity is improving, worsening, or stable. Craniosynostosis generally worsens as the skull growth attempts to compensate for the fused suture. Family histories of similar cranial findings or genetic disorders is pertinent. Developmental milestone achievement should be delineated, noting any signs of delay or regression that may raise concern of increased intracranial pressure secondary to craniosynostosis or other pathology. Other symptoms of increased intracranial pressure can include inconsolability, head holding, drowsiness, or emesis. 1 , 4
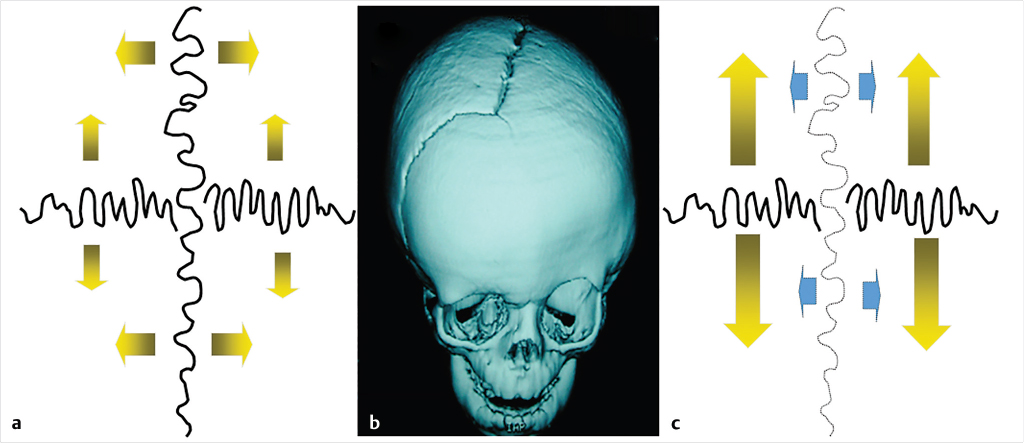
Clinical examination first focuses on general head shape and identifying asymmetries. According to Virchow’s law, early closure of a cranial suture restricts perpendicular growth but causes a compensatory increase of parallel skull growth (Fig. 14‑1). 5 Therefore, characteristic head shapes are often linked with specific craniosynostosis patterns (Fig. 14‑2). However, these associations are not always present, and in some cases, the head shape may appear normal even with multiple fused sutures. 4 , 6 Conversely, the head shape may appear grossly abnormal without any underlying craniosynostosis, such as with positional plagiocephaly. During examination, it is also necessary to palpate sutures and fontanelles. A ridge of bone at a suture line or fontanelle closure may indicate premature suture fusion. An open fontanelle should be soft when an infant is calm and upright. If it is full or tense, this merits consideration of increased intracranial pressure. Additional examination of facial features and extremities should be performed as these may indicate an underlying syndromic diagnosis (Table 14‑1). The patient’s neck should be examined for range of motion to evaluate for contributing torticollis. If exorbitism, eye irritation, or exposure keratitis is noted, prompt referral to an ophthalmologist is needed.

Some patients present with radiographic imaging. Plain radiographs of the skull are of low diagnostic value in this setting, and their results should not overshadow historical and clinical findings. In cases where preoperative imaging is desired, plain radiographs are likely inadequate as well.
14.3 Preparation for Surgery
If the initial consultation yields suspicion of craniosynostosis, referral to a craniofacial surgeon at a tertiary center is necessary, and further consultations and imaging should be considered. Standard consultations at most specialized tertiary centers include neurosurgery and ophthalmology. Neurosurgery will evaluate for cranial pathology and potential surgical assistance, and ophthalmology will evaluate for papilledema or optic atrophy indicative of increased intracranial pressure. Additional consultations may include genetics to assess for a syndromic diagnosis, associated laboratory evaluations, and counseling. If a syndromic diagnosis is suspected or the patient has complex medical issues, a formal craniofacial team evaluation and follow-up is warranted. At tertiary centers, the craniofacial surgeon is often one member of the craniofacial team that will care for complex craniofacial patients. Patients with syndromic associations and complex medical issues are appropriately referred by the team to additional specialists for evaluation and treatment as needed. Preoperative evaluation by an anesthesiologist familiar with pediatric craniofacial patients is performed to ensure perioperative risk is avoided or mitigated as much as possible.
In most cases, preoperative imaging is desired to confirm the diagnosis, identify other abnormal anatomy, such as emissary veins, and assist with overall surgical planning. A computed tomography (CT) scan without contrast is commonly performed, and three-dimensional reconstruction images are obtained. The CT is performed with contrast if vascular drainage is a concern, as in syndromic craniosynostosis. 7 If warranted, magnetic resonance imaging (MRI) may provide additional information about intracranial pathology or posterior fossa malformations, such as tonsillar herniation.
Timing of the operation depends on the severity of the patient’s symptoms. If there are signs of increased intracranial pressure or developmental concerns, surgery proceeds as soon as is feasible. In the absence of increased intracranial pressure, surgery is generally performed within the first year of life, while the exact timing depends on the underlying diagnosis and surgical technique.
Once the surgical strategy and date are determined, patients can be placed on iron supplementation or given recombinant erythropoietin several weeks prior to surgery to optimize hemoglobin levels. 8 Preoperative labs generally include a complete blood count and basic metabolic panel within 30 days prior to surgery.
The most common complications are asymmetry, contour deformities, superficial wound infection, scarring, and wound healing issues. Other risks of any operation for craniosynostosis include bleeding, pain, deep wound infection, blood transfusion, dural tears, cerebrospinal fluid leaks, meningitis, encephalitis, neurologic damage and impairment, and death. These risks should be discussed with caregivers well in advance of surgery.
14.4 Treatment
14.4.1 Indications
Elevated intracranial pressure is an urgent indication for surgical intervention, as it means the brain is externally compressed by the calvarium. In the case of craniosynostosis, the calvarium is unable to accommodate brain growth. The most rapid growth occurs in the first year of life, and the brain nearly triples in size by 2 years of age. 4 Thus, if craniosynostosis is present, as the brain grows the elevated intracranial pressure will likely worsen. Generally, the risk of elevated intracranial pressure increases with each additional suture fusion, and those with syndromic diagnoses have the highest rate of elevated intracranial pressure. 4 , 9 , 10 A craniofacial dysostoses syndrome may be associated with ventriculomegaly, venous outflow obstruction, or obstructive sleep apnea, all of which contribute to elevated intracranial pressure. It is associated with multisuture craniosynostosis over 40% of the time and associated with single suture nonsyndromic craniosynostosis over 15% of the time. 1 , 11 If guardians elect not to proceed with surgery, the patient should be followed up at regular intervals until several years of age to avoid an undiagnosed late presentation of increased intracranial pressure. 6 Untreated increased intracranial pressure can result in neurodevelopmental delay and permanent impairment. 12
Psychosocial development is another indication for surgery. The cranial deformity may result in social dysfunction in both the short and long term. This directly relates to self-image, confidence, and ultimately, quality of life. 13 Therefore, although psychosocial impact may not be an urgent surgical indication, it is a substantial indication for surgery. For these cases, surgery is still generally recommended before 1 year of age to optimize postsurgical ossification.
14.5 Operative Techniques
Surgical interventions for craniosynostosis aim at expanding cranial volume to accommodate the growing brain as well as normalizing overall head shape. The intervention is selected based on patient age, suture synostosis pattern, and associated syndromic deformities. All procedures are performed under general anesthesia with an anesthesiologist familiar with pediatric craniofacial surgery. Blood transfusion reduction strategies, such as tranexamic acid, should be employed where possible. 14 Even with attempts to minimize transfusion rates, surgery should not begin until blood products are available in the operating room.
14.5.1 Strip Craniectomy
A strip craniectomy may be performed through a full coronal approach, partial coronal incision, or multiple endoscopic access incisions (Fig. 14‑3). This technique is most commonly utilized for single suture, nonsyndromic craniosynostosis involving the sagittal suture. For sagittal synostosis, strip craniectomy is most effective in a patient younger than 4 to 6 months and in conjunction with postoperative helmet shaping therapy for 4 to 6 months. Strip craniectomy relies on the malleability of the cranial vault to respond to postoperative helmet therapy. The older the child, the thicker and less malleable the cranial bone, making the child not a candidate for the procedure after 4 to 6 months of age. This makes prompt referral to a craniofacial surgeon imperative if this is to be considered as a treatment option. A craniofacial surgeon and neurosurgeon typically work together to complete the operation, although in some centers the neurosurgeon may perform it alone.
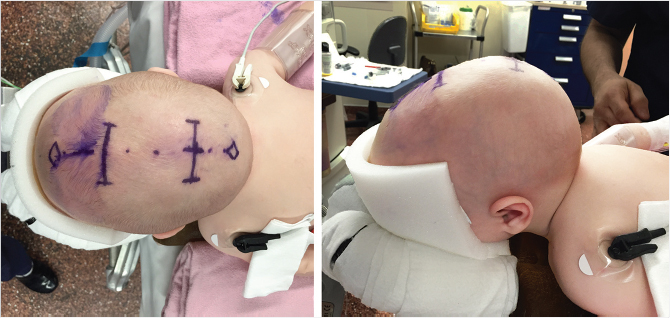
The patient is placed in the supine position on a neurosurgical head rest. Foley catheter, arterial line, and intravenous access are placed. Local anesthetic containing epinephrine is injected along the incision, and then skin is incised. The skin and subcutaneous tissue are undermined as one flap from the periosteum. The area undermined extends a few centimeters beyond the synostotic suture to be removed. The craniectomy is marked. For the sagittal suture, this is approximately 2 to 4 cm in width extending to the anterior and posterior fontanelles. The periosteum is cautiously incised along these markings and undermined from the calvarium. Craniotomy is performed with a burr at the four corners of the desired craniectomy. A curved elevator is gently used to dissect the dura from the inner surface of the calvarium along the craniectomy markings. Extreme caution must be taken near midline as the sagittal sinus may be tethered or partially encased in bone. Rupture of the sagittal sinus can result in significant rapid blood loss that can be fatal. If the sagittal sinus is entered, digital pressure is applied, anesthesia is notified to mobilize blood products, and the cranial vault exposure is widened to access the area of bleeding.
After craniotomy is complete, the craniectomy is performed along the previous markings using a combination of craniotome, Mayo scissors, and endoscope. The piece of bone is slowly removed exposing the sagittal sinus. Barrel stave osteotomies are performed at the left and right sides of the anterior and posterior margins, abutting the fontanelles. These osteotomies are triangular with the apex oriented laterally. With postoperative helmet therapy, these triangular excisions will assist in shortening the anteroposterior length of the cranium and rounding the head shape. The triangles are marked, dissected, and removed in similar fashion to the midline craniectomy. Sealants and coagulative foam are placed along bony margins and the exposed dural surface to assist in hemostasis. Incisions are closed with absorbable suture in a layered fashion. There is no need for a drain, as the dissection is limited.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


