Key points
- •
The human microbiome consists of a wide variety of microorganisms that assist in key physiologic processes.
- •
The majority of microbial diversity is found on the skin and in the gut; and microbial composition is influenced by multiple host factors.
- •
Disruption in microbiome homeostasis is termed dysbiosis and is associated with disease states such as atopic dermatitis.
- •
Flares of atopic dermatitis are associated with changes in the cutaneous microbiome, most notably increases in Staphylococcus aureus and loss of microbial diversity.
- •
The gut microbiome is influenced by multiple factors and evolves over time, with differences observed in patients with atopic dermatitis compared to controls.
Introduction
Interest in the human microbiome and its role in both normal human physiology and in disease has grown dramatically in recent years. Our understanding of the composition and abundance of organisms in the human body has increased as more precise techniques have been developed. No longer viewed as bystanders, commensal organisms are now understood to play vital physiologic roles, including nutrient metabolism, vitamin synthesis, barrier protection, and immune system development. Dysbiosis, a term used to describe imbalance in the healthy microbiome, can lead to increased inflammation and is associated with disease states. This chapter will explore the composition of the healthy microbiome and the changes that occur in dysbiosis and its impact on atopic dermatitis.
Microbiome
The human microbiome refers to the genetic material (collection of all genomes) of the community of organisms, including bacteria, fungi, and viruses, that inhabit the body both inside and out. This adds significant genetic information to the human body, as the amount of nonhuman cells is estimated to be in the trillions and accounts for at least as many human cells, and potentially even 10-fold more ( ). The majority of microbes reside in the intestinal tract, followed next by the skin ( ).
Use of sequencing techniques has led to a better understanding of the composition of the microbiome. These sequencing techniques can either be directed toward a specific genetic target or the genome as a whole ( ). For bacteria, 16 S ribosomal RNA (rRNA) sequencing targets a conserved and yet highly variable region of bacterial genomes, allowing for increased detection of microbes compared to traditional culture techniques ( ). For fungi, sequencing of ribosomal internal transcribed spacer (ITS) allows for identification to the species level ( ).
Whole genome (shotgun) sequencing, a technique used to analyze all genomic material in a given sample, has helped to further classify skin microbes. Compared to 16 S rRNA and ITS sequencing, this technique confers the additional benefits of identifying viral genes, classifying bacterial species to the strain level, and quantifying relative abundances at the kingdom level ( ). Using these techniques, the overall composition of the skin demonstrates highest abundance of bacteria, followed by viruses and lastly fungi (~4%), though site-specific compositional differences exist ( ). Nineteen different phyla of bacteria have been identified on the skin, with the four predominant phyla among these being Actinobacteria, Firmicutes, Proteobacteria, and Bacteroidetes, with the most abundant genera being Corynebacteria, Propionibacteria , and Staphylococci ( ). The most abundant viruses are bacteriophages (viruses that target bacteria), and the predominant fungal genus is Malassezia ( ).
The predominant species found on human skin evolves over time, dependent upon various environmental and physiologic factors. As infants, the microbiome is first influenced by delivery method. Vaginal delivery results in colonization by common vaginal bacteria ( Lactobacillus, Prevotella, Sneathia ), while delivery through cesarean section leads to colonization with common skin bacteria ( Staphylococcus, Corynebacterium, Propionibacterium ) ( ). Regardless of delivery method, this colonization is temporary, with diversification by body site occurring by 6 weeks of age ( ). In prepubescence, the microbiome shifts to an abundance of Firmicutes (including Streptococcaceae ), Bacteroidetes, and Proteobacteria, while changes in hormone levels at puberty result in increased sebum production and subsequent expansion of Propionibacterium, Cornyebacterium , and Malassezia ( ).
Beyond these age-related changes, multiple additional environmental factors have been shown to influence the skin microbiome, including pH, gender, ethnicity, geography, and ultraviolet (UV) exposure ( Table 6.1 ). Healthy skin exhibits a lower skin pH (in the acidic range of 4–6), allowing for the growth of select bacteria (coagulase-negative Staphylococcus, Corynebacteria), while inhibiting pathogenic strains ( Staphylococcus aureus and Streptococcus pyogenes ) ( ). Gender differences also influence the skin microbiome, likely due to inherent differences in hormone, sebum, and sweat production as well as differences in skin surface pH ( ). Numerous studies have also shown variations in the skin microbiome of patients with different ethnic and geographic backgrounds, which likely result from differences in diet and lifestyle among studied populations ( ). For example, Propionibacteriaceae , Staphylococcaceae , and Streptococceacea are more abundant on adult hands in the United States, while Rhodobacteraceae and Nocardioidaceae , bacteria commonly found in soil and aquatic environments, are more abundant on adults’ hands in Tanzania ( ). UV light has an immunomodulatory influence on skin and can decrease skin S. aureus levels ( ). Though influenced by many factors, once established, an individual’s skin microbiome remains stable over time ( ). Factors that influence skin microbiome are listed in Table 6.1 .
|
Skin microbial communities also vary based on body location, which can be categorized by areas that are moist, dry, or sebaceous. Moist areas include the axillae, nostrils, antecubital and popliteal fossae, interdigital web spaces, inguinal creases, plantar heels, and umbilicus (skin creases). These areas tend to have greater abundance of Corynebacterium and Staphylococcus— microorganisms that thrive in environments with higher temperature and humidity. Dry areas include the forearms, palms, and buttocks. These areas tend to have more mixed populations, with an abundance of β-Proteobacteria and Flavobacteriales. Sebaceous areas include the glabella, alar crease, inside and behind the ear, occiput, upper chest, and back; these areas have the lowest amount of diversity, attributed to the anoxic and lipid-rich environment. Sebaceous areas are dominated by Propionibacteria , though Staphylococci are also present ( ). Like bacteria, viruses tend to be less diverse at sebaceous sites ( ). While Malassezia is the predominant fungus on the majority of skin surfaces, increased fungal diversity is seen on parts of the foot ( ).
In its normal state, the skin microbiome is in a healthy homeostasis and consists primarily of commensal organisms, or those conferring benefit without causing harm, and transient microbes ( ). This homeostasis is maintained by a combination of protective mechanisms inherent to the skin, as well as contributions from a diverse resident bacterial population. Keratinocytes produce numerous antimicrobial peptides (AMPs), including cathelicidin and β-defensin, that protect the skin barrier through multiple mechanisms, including direct attack on microbes and eliciting a host response ( ).
Similar to innate host defense, commensal bacteria ( Staphylococcus epidermidis being the most widely studied) also contribute to this balance through multiple mechanisms. Directly, S. epidermidis can produce peptides that inhibit growth of pathogenic organisms such as S. aureus , without disrupting other commensals ( ). Indirectly, S. epidermidis is capable of eliciting an immune response targeting pathogens through activation of toll-like receptors ( ). S. epidermidis has also been shown to inhibit the formation of S. aureus biofilms ( ).
Regulatory T cells (Tregs), a subset of CD4+ T cells, play an important role in immune regulation and maintain-ing homeostasis through promotion of tolerance to self-antigens. A greater number of regulatory T cells are present in infancy ( ), and exposure to commensal bacteria on the skin early on in life plays a significant role in establishing this tolerance ( ). Specifically, in the skin, the early presence of S. epidermidis is detected by antigen-presenting cells, presented to T cells that express the matching antigen receptor, which results in the expansion of a population of regulatory T cells that allows for tolerance to the organism ( ). As a result, subsequent detection of S. epidermidis does not trigger inflammation (immune tolerance), but instead results in the induction of cytokine interleukin 1a (IL1a), which in turn acts as host defense, an example of immune-commensal crosstalk ( ). Early exposure is necessary for this commensal tolerance to occur ( ). Since regulatory T cells play a key role in self-tolerance, dysfunctional or decreased number of Tregs are seen with autoimmune conditions ( ).
Skin dysbiosis
Dysbiosis refers to a disruption of the microbiome and a shift away from homeostasis, and is seen in disease states such as atopic dermatitis. A study published in 1989 highlighted epidemiologic data that correlated increases in the incidence of atopic dermatitis with improved personal cleanliness and smaller family sizes (with subsequent decreases in sibling contact), suggesting that a disruption in microbial exposure may contribute to atopic dermatitis ( ). This idea became known as the hygiene hypothesis. In support of this hypothesis were subsequent data that showed that exposure of children to microbial products (endotoxin) through their environment was protective against the development of atopic disease ( ).
Risk factors of skin dysbiosis
Patients with atopic dermatitis are at increased risk for dysbiosis through skin barrier dysfunction ( Table 6.2 ). Extrinsically, as flares drive itch, scratching physically breeches the barrier. Intrinsic factors include the higher skin pH seen in these patients, which results in altered synthesis of protective lipids that creates an environment conducive to pathogen growth, including Staphylococcus and Candida ( ). Loss-of-function mutations in filaggrin, a protein with multiple roles important to skin barrier function, including pH regulation and maintaining hydration, is a major risk factor for the development of atopic dermatitis and is present in nearly half of patients ( ).
| Barrier Dysfunction |
|---|
| Defective lipid matrix |
| Decreased expression of tight junction proteins |
| Increased production of proteases |
| Scratching |
| Filaggrin mutation |
| Increased water loss |
| Decreased hydration |
| Increased pH |
| Microbiome |
| Decreased diversity |
| Increased Staphylococcus aureus |
| Immune Dysregulation |
| Increased inflammation |
| Increased Th2 response |
| Allergic sensitization |
| Decreased antimicrobial peptides |
| Impaired innate and adaptive immune response |
Patients with atopic dermatitis also produce fewer antimicrobial peptides, such as cathelicidin and β-defensins, predisposing individuals to increased S. aureus colonization ( ). Antibiotics, commonly used as part of the treatment for atopic dermatitis, may be an additional contributor to dysbiosis through nonselective elimination of both beneficial and pathogenic bacteria ( ). Together, these factors contribute to a state of dysbiosis by altering the skin’s natural defenses and allowing allergens and microbes to enter the skin (a state also known as leaky skin), ultimately resulting in inflammation ( ).
Bacterial composition
In a healthy state, coagulase-negative staphylococci (CoNS) species such as S. epidermidis, Staphylococcus lugdunensis , and Staphylococcus caprae limit S. aureus colonization and pathogenicity ( ). In addition to CoNS, additional microbes beneficial to maintaining homeostasis have been identified. Propionibacterium and Corynebacterium may also limit growth of S. aureus through their ability to release porphyrins ( ). Streptococcus pneumoniae is capable of killing S. aureus through release of hydrogen peroxide ( ). Roseomonas mucosa , a gram-negative bacterium, has also been shown to limit S. aureus growth and influence innate immune activation ( ).
A shift to dysbiosis is marked by low bacterial diversity in general, though with higher levels of staphylococcal species ( S. aureus and S. epidermidis ), and a greater diversity of non- Malassezia fungi ( ). Since the 1970s, S. aureus has been known to colonize the skin in 30% to 100% of atopic dermatitis patients, in contrast to approximately 20% of those unaffected, and to contribute to atopic dermatitis flares ( ). Atopic dermatitis patients are particularly at risk for this colonization and subsequent dysbiosis due to reduced levels of antimicrobial peptides, a consequence of both suppression from increased Th2 cytokines and decreased coagulase-negative staphylococcal species ( ).
Importance of Staphylococcus aureus
Virulence of S. aureus starts with its ability to adhere to and penetrate atopic dermatitis skin ( ) and is achieved through the expression of multiple proteins. The toxins produced by S. aureus can be categorized into three general groups based on function: toxins that damage cell membrane (which includes α- and δ-toxins), toxins that interfere with receptor function (which includes the superantigen enterotoxin), and secreted enzymes (proteases that alter host pathways) ( ). Atopic dermatitis patients often exhibit reduced or impaired filaggrin expression, making these patients more susceptible to S. aureus toxins. Reduced filaggrin expression leads to reduced sphingomyelinase secretion, an enzyme that hydrolyzes sphingomyelin into ceramides, and which has been shown to protect keratinocytes specifically from α-toxin and cell death ( ). δ-Toxin has also been shown to cause mast cell degranulation, promote IL4 production, and lead to skin inflammation, as seen in atopic dermatitis ( ). Numerous other proteins produced by S. aureus may contribute to atopic dermatitis, including those that allow adhesion to skin (clumping factor B, fibronectin-binding proteins), proinflammatory proteins (protein A, lipoproteins, phenol-soluble modulins), and those that inactivate AMPs (staphopain, aureolysin) ( ). The ability to form biofilms also helps protect S. aureus from host immune response and antibacterial treatment ( ).
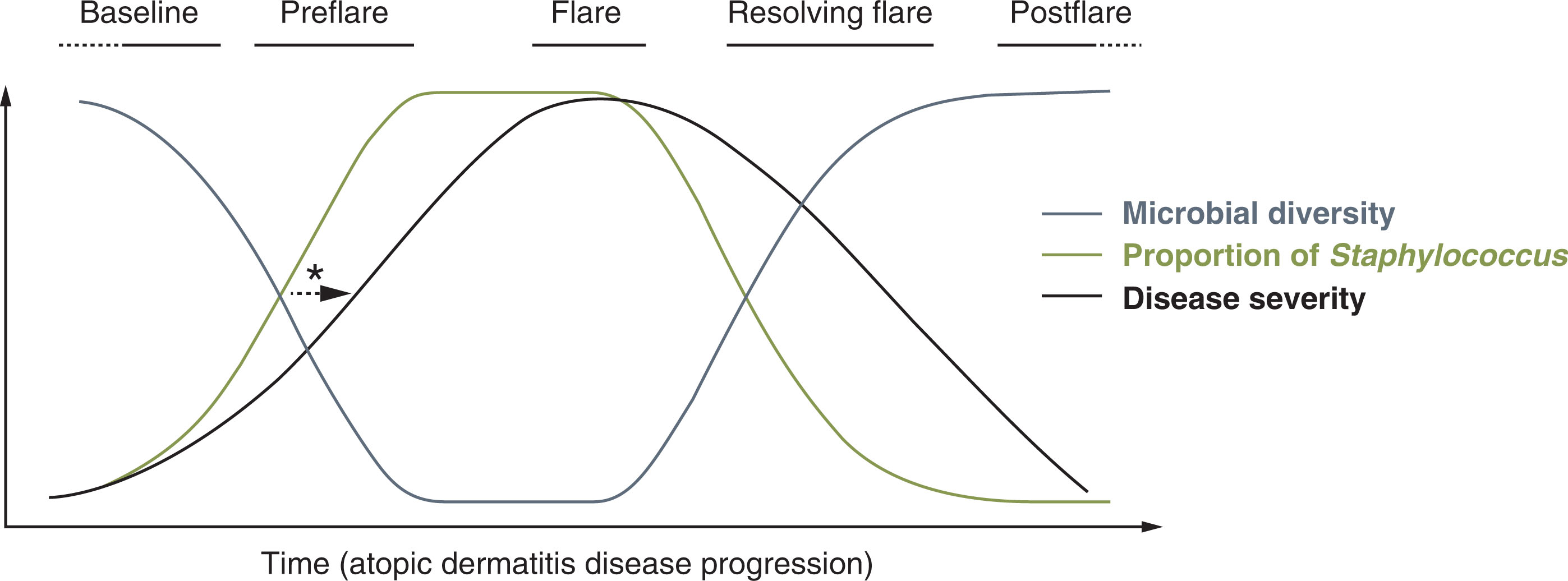
S. aureus density is increased in lesional skin of atopic dermatitis patients and correlates with its disease severity. During a disease flare, and even preceding the flare, higher levels of S. aureus and an overall decrease in microbial diversity are observed. As the flare resolves, levels of S. aureus fall, and microbial diversity is restored ( ) ( Fig. 6.1 ). It has also been observed that atopic dermatitis patients experiencing severe flares are colonized not only with S. aureus , but also with other strains that have great capability of inducing an inflammatory response, while those with less severe atopic dermatitis have a predominance of S. epidermidis ( ). Following treatment of flares, the microbiome shifts, with a decrease of Staphylococcus species and increases in Streptococcus, Corynebacterium , and Propionibacterium species ( ).
Other factors
Although S. aureus is the most widely implicated and studied microbial contributor to dysbiosis in atopic dermatitis, other microbes such as Propionibacterium, Streptococcus, Acinetobacter , and Malassezia are also believed to play a role ( ). Among other observations, lesion-prone skin of atopic dermatitis patients is higher in Streptococcus and Gemella species but lower in Dermacoccus , a genus of bacteria capable of producing antiinflammatory and antimicrobial metabolites ( ). Although Malassezia is considered part of the normal skin flora, as many as 80% of patients with atopic dermatitis are sensitized to and produce immunoglobulin E (IgE) against Malassezia , a response that drives inflammation ( ). It has also been shown that a specific fungal protein from Malassezia (MGL_1304 from Malassezia globosa ) is found in human sweat and causes histamine release from basophils, with increased levels of IgE against this protein seen in atopic dermatitis patients during flares and correlating with severity ( ). Shifts in predominant Malassezia species (specifically Malassezia restricta and M. globosa ) also occur, based on severity of disease. Though marked by a lower bacterial diversity, patients with atopic dermatitis have a higher diversity of fungi—specifically non- Malassezia fungi—when compared to controls, including the presence of Candida and Cryptococcus species ( ).
Additional factors found to support a diverse microbiome include emollient use ( ) and treatment of atopic dermatitis ( ). However, treatments with antibacterial agents alone, aimed specifically toward reduction of S. aureus , do not seem to improve atopic dermatitis flares in the absence of clinical infection ( ). For example, though used widely in clinical practice as a means of decreasing S. aureus burden, current evidence suggests that bleach baths are too diluted to be bactericidal ( ) and that their use may confer no additional benefit over water bath alone ( ). Together, these findings suggest a greater interplay among many factors in the connection between dysbiosis and atopic dermatitis.
Gut dysbiosis
The gut microbiome is composed of trillions of microbes and is postulated to assist crucial physiologic functions such as nutrient and protein metabolism, vitamin synthesis, drug metabolism, and protection of the gut epithelial barrier ( ). It also plays a key role in the development of the early immune system. Though the womb was once considered sterile, colonization of the gut is now believed to begin in utero, with maternal bacteria identified in umbilical cord blood, amniotic fluid, meconium, and the placenta ( ). Similar to the evolution seen on the skin, the gut microbiome also changes over time. The gut of neonates is briefly dominated by an abundance of Enterobacteriaceae and Staphylococcus before then evolving to a Bifidobacterium -predominant environment while breastfeeding. Once started on solid foods, the gut microbiome shifts to a predominance of Bacteroidetes and Firmicutes ( ).
Establishment of the gut microbiome
Factors influencing the initial establishment and composition of the gut microbiome in infancy include timing of birth, delivery method, feeding method, and antibiotic use ( Table 6.3 ). Preterm infants have decreased levels of Bifidobacterium and are at risk for further alterations in gut flora due to subsequent parenteral nutrition and antibiotic use ( ). Vaginal delivery is associated with increased Bifidobacterium and Bacteroides species, while delivery by cesarean section is associated with higher numbers of Streptococcus, Staphylococcus , and Clostridium difficile ( ). This influence of delivery method on the gut microbiome may even carry into adulthood ( ). Infants born by cesarean section are at increased risk of development of atopic dermatitis compared to those born via vaginal delivery ( ). Infants that are exclusively breastfed show increases in the class Actinobacteria (includes genus Bifidobacteria ), while formula-fed infants have higher levels of Proteobacteria (which includes the genus Escherichia ) ( ). Though results are mixed, many studies have shown that breastfed infants are also at a decreased risk of developing atopic dermatitis, particularly those with a family history of atopy, with the American Academy of Allergy, Asthma, and Immunology recommending exclusive breastfeeding for 4 to 6 months to reduce this incidence ( ).
| Timing of Birth | |||
|---|---|---|---|
| Preterm | ↑ | Clostridium difficile | ( ) |
| ↑ | Proteobacteria | ( ) | |
| ↑ | Enterococcus | ( ) | |
| Delivery Method | |||
| Cesarean section | ↑ | C. difficile | ( ) |
| ↑ | Klebsiella | ( ) | |
| ↑ | Veillonella | ( ) | |
| ↑ | Streptococcus | ( ) | |
| ↑ | Staphylococcus | ( ) | |
| ↑ | Cornyebacterium | ( ) | |
| ↑ | Propionibacterium | ( ) | |
| ↓ | Bifidobacterium | ( ) | |
| ↓ | Bacteroides | ( ) | |
| ↓ | Lactobacillus | ( ) | |
| Vaginal delivery | ↑ | Bifidobacterium | ( ) |
| ↑ | Lactobacillus | ( ) | |
| ↑ | Prevotella | ( ) | |
| ↑ | Sneathia | ( ) | |
| Feeding Method | |||
| Formula | ↑ | C. difficile | ( ) |
| ↑ | Proteobacteria | ( ) | |
| Breastfeeding | ↑ | Bifidobacterium | ( ) |
| ↑ | Actinobacteria | ( ) | |
| ↓ | Escherichia coli | ( ) | |
| ↓ | C. difficile | ( ) | |
| ↓ | Bacteroides fragilis | ( ) | |
| Infant Antibiotic Exposure | |||
| Direct | ↑ | Enterobacteriaceae | ( ) |
| ↓ | Bifidobacterium | ( ) | |
| ↓ | Bacteroides | ( ) | |
| ↓ | Lactobacillus | ( ) | |
| ↓ | Lachnoclostridium | ( ) | |
| Maternal (peripartum) | ↑ | Enterobacteriaceae | ( ) |
| ↓ | Bacteroides | ( ) | |
| ↓ | Bifidobacteria | ( ) | |
| Maternal Weight | |||
| Maternal obesity | ↑ | Clostridium histolyticum | ( ) |
| ↓ | Bacteroides-Prevotella | ( ) | |
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


