Abstract
The clinical differential diagnosis of “hard skin” can be divided into two main diagnostic categories: morphea and morphea-like conditions and systemic sclerosis and sclerodermoid conditions. Patients with morphea and morphea-like conditions tend to have more asymmetric, discontinuous skin involvement, while patients with systemic sclerosis and sclerodermoid conditions tend to have more symmetric, distal, and continuous skin involvement. Although morphea and systemic sclerosis share a common endpoint of “hard skin” with a histologic correlate of cutaneous sclerosis, differences in pathophysiology suggest they are distinct disease processes rather than a disease spectrum. Further, while morphea tends to be localized to the skin and structures directly underlying, systemic sclerosis demonstrates the highest disease-related mortality of any autoimmune connective tissue disease due to visceral involvement (e.g., interstitial lung disease and pulmonary arterial hypertension). From a practical standpoint, the presence of sclerodactyly, nailfold capillary changes, and Raynaud’s phenomenon are useful in distinguishing systemic sclerosis and its attendant complications from other causes of hard skin.
Keywords
Eosinophilic fasciitis, Mixed connective tissue disease, Morphea, Nephrogenic systemic fibrosis, Raynaud’s, Scleroderma, Systemic sclerosis
- •
The range of conditions presenting as “hard skin” is broad, including those diseases that cause cutaneous sclerosis (increased connective tissue with normal or decreased fibroblasts) and those that cause cutaneous fibrosis (increased connective tissue and increased fibroblasts).
- •
The clinical differential diagnosis of “hard skin” can be divided into two main categories: morphea and morphea-like conditions and systemic sclerosis and sclerodermoid conditions.
- •
Patients with morphea and morphea-like conditions tend to have more asymmetric, discontinuous skin involvement, while patients with systemic sclerosis and sclerodermoid conditions tend to have more symmetric, distal, and continuous skin involvement.
- •
The presence of sclerodactyly, nailfold capillary changes, and Raynaud’s phenomenon are useful in distinguishing systemic sclerosis from other causes of hard skin.
Scleroderma
The term “scleroderma” is frequently used in reference to both localized (morphea) and systemic (systemic sclerosis) conditions presenting with “hard skin.” While both morphea and systemic sclerosis (SSc) share a common endpoint of cutaneous sclerosis with identical histologic features, key differences in their pathophysiology, autoantibody profile, and clinical presentation suggest they represent separate disease processes rather than a spectrum. Importantly, morphea does not evolve into SSc, and patients with morphea do not develop the specific internal organ manifestations of SSc. As a general rule, patients with morphea tend to have more asymmetric, discontinuous skin involvement, and patients with SSc tend to have more symmetric, distal, and continuous skin involvement. Thus, the differential diagnosis of “hard skin” can be separated into two main diagnostic categories: morphea and morphea-like conditions and systemic sclerosis and sclerodermoid conditions.
Morphea (Localized Scleroderma)
Clinical Manifestations
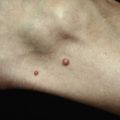
Morphea (localized scleroderma) occurs in both adults and children and may present as one or more edematous, indurated, or atrophic plaques. Morphea is typically erythematous or violaceous in its active (or inflammatory) phase ( Fig. 3-1 ) and ivory-colored or hyperpigmented in its damage (or noninflammatory) phase. Morphea is distinguished from SSc based on the absence of sclerodactyly, Raynaud’s phenomenon, and nailfold capillary changes. The term “morphea” may be preferred over “localized scleroderma” to emphasize the distinction from SSc and its specific end-organ complications. Extracutaneous manifestations of morphea can include involvement of underlying structures such as bone and, in cases of linear morphea affecting the head, the central nervous system. Arthralgias may also be present (e.g., 10% of pediatric patients) ( ). ANA (antinuclear antibody) positivity is observed in 20% to 80% of patients with morphea, but is generally not indicative of an underlying systemic autoimmune connective tissue disease such as lupus erythematosus or SSc.
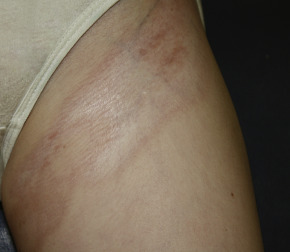
While the classification of morphea subtypes has been controversial, one of the more commonly used systems proposed by Laxer and Zulian describes five morphea variants based on clinical manifestations: circumscribed, linear, generalized, pansclerotic, and mixed variants. Circumscribed morphea, the most common subtype in adults, presents with up to three individual plaques. Active lesions often demonstrate a characteristic indurated lilac rim ( Fig. 3-1 ); they may expand or burn out, becoming ivory-white or hyperpigmented and softening over several years. Both superficial (more common; limited to the epidermis and dermis) and deep (involving the deep dermis and subcutaneous tissues) variants of circumscribed morphea have been described.
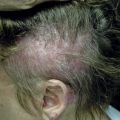
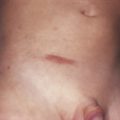
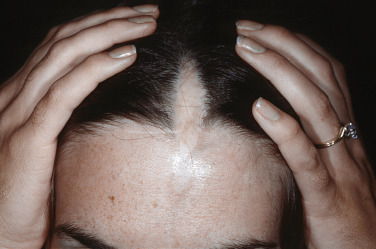
Linear morphea presents as a band of sclerotic, depressed skin, frequently with overlying hyperpigmentation. Linear morphea is most common in children and affects the limbs, face, and/or scalp, typically as a single lesion ( Fig. 3-2 ). In children, linear morphea of the limbs can cause focal growth arrest and limb length discrepancies due to disruption of the growth plate. Joint contractures are a particular concern when the lesion crosses a joint. The head variant of linear morphea includes two subtypes: en coup de sabre (ECDS; frontoparietal linear morphea) and progressive hemifacial atrophy (PHA; Parry Romberg syndrome). En coup de sabre ( Fig. 3-3 ) most often presents as a depressed and/or hyperpigmented plaque on the paramedian forehead; when the lesion extends into the scalp, prominent alopecia may be observed. Early in its course, ECDS can present as an erythematous patch mimicking a port wine stain. In contrast, PHA predominantly affects the subcutaneous tissues, sometimes with only subtle changes of overlying skin. Overlapping features of both ECDS and PHA in the same patient are not uncommon. Head variant linear morphea is associated with central nervous system and eye abnormalities. Central nervous system manifestations include seizures and headaches, reported in 13% and 9% of patients, respectively. The severity of skin findings is not predictive of CNS abnormalities, and a magnetic resonance image (MRI) with contrast is helpful to guide management. Ocular manifestations may be present in 3% of patients with head variant linear morphea, with common features including adnexal sclerosis and uveitis; detection by serial ophthalmologic examinations may help prevent permanent visual loss. Dental abnormalities may also be present, particularly in patients with PHA. Although most common in children, linear morphea may also present in adults.
Generalized morphea describes the presentation of four or more indurated plaques, larger than 3 cm each, involving two or more separate anatomical areas ( Fig. 3-4 ). Arthralgias may be more prevalent than in other morphea subtypes. Patients with generalized morphea are more likely to have positive autoantibodies. Severe generalized morphea can be distinguished from SSc based on the absence of Raynaud’s phenomenon, nailfold capillary changes, and sclerodactyly.
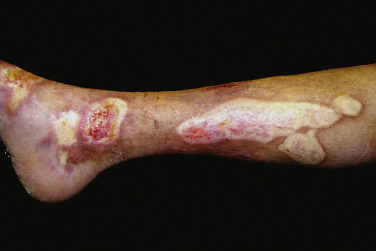
Pansclerotic morphea is a poorly defined, very rare, and aggressive morphea subtype, which has been proposed by some authors to be a variant of generalized morphea. Pediatric patients in particular may have circumferential and subcutaneous involvement, affecting nearly the entire body surface area. Pansclerotic morphea may also present with full-thickness involvement of the underlying muscle, tendon, and bone, with associated nonhealing ulcers and cutaneous squamous cell carcinoma.
Up to 15% of morphea patients present with a combination of two or more of the previously discussed subtypes, or the “mixed subtype” variant. Other entities that may fall along the morphea spectrum include lichen sclerosus and eosinophilic fasciitis (discussed separately later in this chapter). Genital lichen sclerosus has been reported to occur with greater than average frequency in individuals with plaque, linear, and generalized morphea; a genital exam is ideally included in the care of patients with morphea. Whether lichen sclerosus truly falls along the morphea spectrum ( Fig. 3-5 ) or is an associated condition requires further clarification.
Diagnosis
The diagnostic approach to morphea includes characterization of the extent and location of involvement as well as exclusion of other common morphea mimics or morpheaform disorders ( Table 3-1 ). Morphea is generally a clinical diagnosis, and skin biopsy may not be required in typical cases. Morphea and SSc are indistinguishable on biopsy. Skin biopsy may nevertheless be useful when atypical findings are present and/or to assess the severity of active inflammation when considering treatment options. Biopsy specimens from well-developed lesions should include underlying subcutaneous tissue and are square in appearance histologically (the so-called “square biopsy”). The epidermis may be normal or atrophic, with thickened and densely packed collagen bundles in the dermis, atrophy or absence of the skin appendages, adnexal trapping, and replacement of the fat cells in the subcutaneous tissue by hyalinized collagen bundles. In the early inflammatory stages of morphea, a predominantly lymphocytic infiltrate, with or without plasma cells, is seen in the dermis and superficial subcutaneous fat ( Fig. 3-6 ).
| Morphea Group | Morpheaform Conditions |
|---|---|
|
|
Differential Diagnosis
The differential diagnosis of morphea includes a number of morpheaform conditions or morphea mimics, some of the most clinically relevant including radiation-induced morphea, cutaneous malignancy, injection site reactions, and lipodermatosclerosis ( Table 3-1 ).
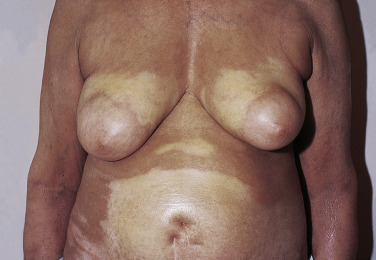
Radiation-induced morphea may be a subtype of “true” morphea triggered by radiation-related immunologic aberration in the affected skin. Radiation-induced morphea typically presents within 1 year after completion of radiation treatment, though delayed presentations can occur. Radiation-induced morphea is most common in women following radiation for breast cancer, where it presents with shrinkage of the affected breast, distinguishing it from radiation dermatitis. In this clinical scenario, biopsy is indicated to rule out recurrent malignancy, primary malignancy (e.g., basal cell carcinoma), and metastatic disease (e.g., carcinoma en cuirasse).
Injection-site reactions and lipodermatosclerosis may also mimic morphea. Cutaneous sclerosis at injection sites has been reported with vitamin B 12 and vitamin K injections, vaccinations, and other injectables. Lesions tend to slowly resolve without treatment. Lipodermatosclerosis, or sclerosing panniculitis, is a manifestation of venous insufficiency identifiable by its typical features of induration and hyperpigmentation along the lower medial aspect of the leg, with a classic “inverted champagne bottle” appearance. Findings may be either unilateral or bilateral and can occasionally simulate morphea or eosinophilic fasciitis. Lipodermatosclerosis is often a clinical diagnosis; biopsy may be deferred in typical cases due to compromised healing of affected regions.
Management
Several treatment algorithms for morphea have recently been proposed. Treatment for morphea is generally tailored to the condition’s activity, severity, subtype, and potential functional and cosmetic implications. Of note, there is little evidence for the treatment of inactive morphea. Some treatment considerations with respect to disease subtype are as follows.
Circumscribed Morphea
Topical corticosteroid monotherapy is a common initial approach to treating circumscribed morphea in practice; while topical corticosteroids alone may indeed be beneficial, there are presently no data to specifically support this. Topical tacrolimus resulted in improved skin thickness in a small randomized controlled trial and is another reasonable approach. If lesions are unresponsive, other options include topical imiquimod, topical calcipotriene, a combination of calcipotriol and betamethasone dipropionate, or lesion-limited phototherapy (NB-UVB, UVA, or UVA-1). The accessibility and limited toxicity of NB-UVB make it a favorable initial form of phototherapy for many patients. Patients with deeper or recalcitrant disease may benefit from a UVA-based regimen—excimer laser.
Linear Morphea
Both prospective and retrospective data support the efficacy of methotrexate (e.g., 15 to 25 mg/week in adults, 1 mg/kg per week in children) in combination with systemic steroids (e.g., prednisone 1 mg/kg PO daily or IV pulse with or without subsequent oral prednisone taper) in the treatment of morphea ( ). In children with linear morphea, methotrexate plus systemic corticosteroids is the standard of care and is supported by data from a randomized placebo-controlled trial. If there is no improvement after 8 to 12 weeks, addition of or transition to phototherapy (UVA1 or PUVA for deeper lesions and a trial of NB-UVB for superficial lesions) or mycophenolate mofetil can be pursued. Pediatric rheumatologists frequently add mycophenolate mofetil in addition to methotrexate in refractory cases. Notably, assessing treatment response can be very challenging in cases of linear morphea. Appropriate counseling is critical in that the first goal of therapy is to halt disease progression; tissue that has already been damaged will not return to normal and should not lead to increasing the patient’s treatment regimen. Reconstructive surgery can be considered for disfiguring facial atrophy, provided the disease has been quiescent for several years. Surgical intervention during active disease has caused rapid progression in individual patients.
Generalized Morphea
Phototherapy is typically considered first-line treatment for generalized morphea if the patient does not have functional limitations. If there is no response after 8 weeks, a combination of methotrexate and systemic corticosteroids can be implemented. If there is no improvement after an additional 8 to 12 weeks, mycophenolate mofetil can be considered.
Systemic Sclerosis
Clinical Manifestations
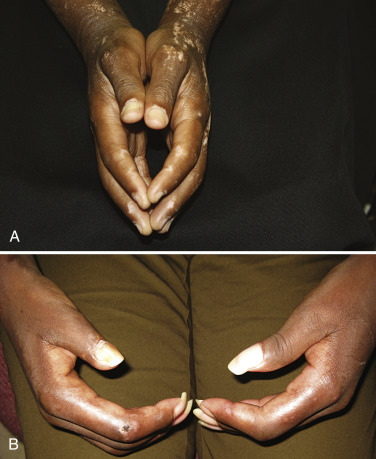
Systemic sclerosis (SSc) is an autoimmune connective tissue disease characterized by vascular dysfunction, autoimmunity, and sclerosis of end organs including skin. While SSc is most prevalent in middle-aged women, it can occur at all ages and in both genders. The etiopathogenesis likely involves both genetic and environmental factors. Disease subtypes include: (1) limited cutaneous systemic sclerosis (formerly known as CREST, i.e., calcinosis, Raynaud’s, esophageal dysmotility, sclerodactyly, telangiectasia); (2) diffuse cutaneous systemic sclerosis; (3) overlap syndromes, including mixed connective tissue disease (discussed separately at the end of this chapter); and (4) systemic sclerosis sine scleroderma (lacking skin involvement).
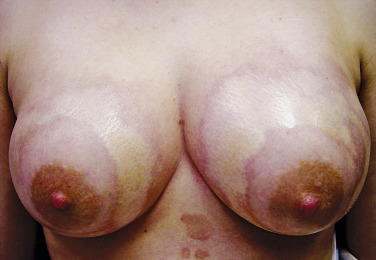
Diffuse cutaneous systemic sclerosis (dSSc) and limited cutaneous systemic sclerosis (lSSc) are the most well-recognized clinical patterns of disease, differentiated based on the extent of skin involvement. Specifically, dSSc is defined by cutaneous sclerosis proximal to the elbows while lSSc is characterized by skin involvement only distal to the elbows and often limited to the most distal extremities. Early disease is characterized by an edematous phase in which patients may present with “puffy fingers” or “puffy hands.” As the disease progresses, the areas of swelling become indurated, with findings progressing from distal to proximal ( Fig. 3-7 ). In dSSc, hand and finger edema may present shortly after the first episode of Raynaud’s phenomenon. In contrast, patients with lSSc may experience Raynaud’s phenomenon for years before other manifestations develop, reflecting what is often a more protracted disease course.