Abstract
Mastocytosis may arise in utero until later in life. Most patients with mastocytosis are children, but the true incidence of adult-onset disease is unknown. While the cause of this disorder remains unknown, auto-activating mutations of c-KIT are frequently detected in both children and adults with mastocytosis. Seven different disease categories of mastocytosis are recognized by the World Health Organization. Most children have cutaneous only disease, while many adults have cutaneous or indolent systemic mastocytosis. Recent studies suggest that adults with more aggressive forms of systemic mastocytosis express additional gene mutations, thus differentiating them from patients with milder disease. Urticaria pigmentosa lesions are usually seen in children, whereas most adults with milder forms of systemic mastocytosis have 2-to 4-mm, red-brown papules with overlying telangiectases. Patients with mastocytosis may have few, if any, symptoms, but when they occur, most are related to mast cell histamine release. Patients with more advanced disease frequently have more symptoms, fewer, if any, skin lesions, and evidence of other organ (gastrointestinal, liver, bone, bone marrow) involvement. The prognosis for patients with cutaneous and limited systemic mastocytosis is very good. Treatment is limited to blocking the effects of released mast cell mediators with antihistamines and leukotriene inhibitors. Tyrosine kinase inhibitors have not proven effective for the treatment of mastocytosis. Omalizumab, which is an anti-IgE monoclonal antibody, also has been reported to control the symptoms, but not the growth of mast cells, in mastocytosis.
Keywords
c-KIT mutations, Histamine, Mast cell disease, Mastocytosis, Urticaria pigmentosa
- •
Mastocytosis is a disease of both children and adults.
- •
Most children have skin-limited disease and an excellent prognosis.
- •
Many adults have either cutaneous only or indolent systemic mastocytosis, and a very good prognosis.
- •
Patients with more advanced disease have a worse prognosis, which may include the development of a second hematologic malignancy.
- •
The diagnosis is established by demonstrating increased mast cells in the skin or other organs.
- •
Treatment is limited to controlling the symptoms of released mast cell mediators.
Mast cell disease, or mastocytosis, represents a spectrum of clinical disorders that results from an abnormal proliferation of mast cells (MCs). The onset ranges from the time of birth to late adulthood. In a report of 101 children with mastocytosis, disease onset occurred in 73% of patients within 6 months of age and 97% of patients by age 2 years. The prevalence of mastocytosis among infants and children ranges from 5.4 cases per 1000 in a Spanish population to 1 case per 500 in a Mexican cohort. The prevalence of mastocytosis in adults is unknown but believed to be even more rare. This disorder is equally distributed between males and females; it has been reported in all races, and most patients have no familial association; however, there have been over 40 cases of familial mast cell disease, some of which have involved several generations.
Pathogenesis
MCs are derived from CD34 + precursor cells arising in the bone marrow and circulate as monocytic cells. Circulating mast cell precursors in the blood express CD34, the tyrosine kinase KIT (CD117), and FcγRII, but not high-affinity IgE receptors (FcεRI). KIT is a type III tyrosine kinase that is expressed on MCs, melanocytes, primitive hematopoietic stem cells, primordial germ cells, and interstitial cells of Cajal. Activation of KIT induces cellular growth and extends cell survival by preventing apoptosis. The ligand for KIT is stem cell factor (SCF), which is important for mast cell growth. SCF is produced by bone marrow stromal cells, fibroblasts, keratinocytes, endothelial cells, and reproductive Sertoli and granulosa cells. Under normal conditions, once mast cell precursors enter tissues they become KIT + /CD34 − /FcγRII − /FcεRI + and develop characteristic cytoplasmic granules.
Alterations in c-KIT structure have been implicated in the pathogenesis of mast cell disease. Specifically, somatic mutations in codon 816 of the c-KIT proto-oncogene have been identified in adults and children with mastocytosis. The result is a substitution of the amino acid aspartic acid (D) with valine (V) or another amino acid; examples include D816V, D816Y, D816F, D816I, and D816H. This mutation causes constitutive activation of KIT, thereby leading to continued mast cell development. Additional mutations in c-KIT (del419, K509I, F522C, V533D, A533D, V559A, V560G, R815K, I817V, D820G, E839K) also have been reported in adults and children with mastocytosis, but are more rare. Frequently these mutations are detected in mRNA not DNA. In a recent study of 50 children with mastocytosis, ranging in age from 0 to 16 years, activating mutations were detected in 86% of the patients. Forty-two percent were in codon 816 and the remainder were within the gene that encodes the proximal extracellular region (fifth immunoglobulin-like domain). All the c-KIT mutations except M541L found in these children were associated with constitutive KIT activation. Interestingly, an inactivating mutation (e.g., E839K) also has been described in a child with mastocytosis, In the extremely rare familial mast cell disease, c-KIT mutation detection has been variable, ranging from none to the expression of K509I and A533D mutations. The finding of inactivating or no c-KIT mutations in some patients and families with mastocytosis suggests that factors beyond an abnormal KIT receptor play a role in this disease. In fact, additional gene mutations have been identified in some adult patients with more advanced mastocytosis and include the tumor suppressor gene TET2 as well as ASCL1 , JAK2 , SRSF2 , DNMT3A , RUNX1 , and CBL . Taken together, these observations suggest that mutations in c – KIT lead to the development of mastocytosis but that additional gene mutations occur and may be necessary for life-long disease.
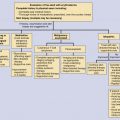
Classification of Mast Cell Disease
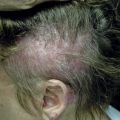
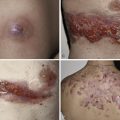
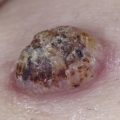
The classification of mast cell disease has been defined by the World Health Organization (WHO) with the following disease categories: cutaneous mastocytosis (CM), indolent systemic mastocytosis (ISM), smoldering systemic mastocytosis (SSM), isolated bone marrow mastocytosis (IBMM), systemic mastocytosis with an associated clonal hematologic nonmast cell lineage disease (SM-AHNMD), aggressive systemic mastocytosis (ASM), mast cell leukemia (MCL), mast cell sarcoma, and extracutaneous mastocytoma ( Table 42-1 ). Patients with CM and ISM represent the largest groups, and include most children with CM and many adults with CM or ISM. All CM patients and many ISM patients have cutaneous lesions. Cutaneous involvement in mast cell disease is defined by typical lesions of mastocytosis (see “Clinical Manifestations”), and pathological changes that demonstrate either monomorphic mast cell clusters (>15 MCs/cluster) or scattered MCs at more than 20 MCs/high-power field (hpf). Systemic mast cell disease is defined by major and minor criteria in which the major criteria are represented by multifocal dense mast cell infiltrates (15 MCs/aggregate) in the bone marrow or other extracutaneous organs, and the minor criteria include the presence of >25% spindle-shaped or atypical-appearing MCs in tissue sections or a bone marrow aspirate smear, the presence of a c-KIT mutation at codon 816, the expression of CD2 and/or CD25 by MCs and a persistent total serum tryptase level of >20 ng/mL ( Table 42-2 ). The diagnosis of SM is established in patients having the major and one minor criteria or three minor criteria. Whereas most patients with SM have indolent disease, a smaller subset has been recognized as SSM because of greater disease burden, e.g., hepatosplenomegaly, lymphadenopathy, serum tryptase levels >200 ng/mL. Another recognized group of SM patients is one with an associated clonal hematologic nonmast cell disorder. Hematologic diseases associated with this SM group include: polycythemia rubra vera, myelodysplastic syndrome, chronic eosinophilic leukemia, chronic myeloid leukemia, chronic myelomonocytic leukemia, lymphocytic leukemia, acute erythroblastic leukemia, megaloblastic leukemia, and non-Hodgkin’s lymphoma. Patients with SM-AHNMD may or may not have skin lesions, but frequently have liver, spleen, and/or lymph node involvement. They often are older adults, and many have constitutional symptoms such as fever, anorexia, weight loss, and generalized malaise. Aggressive systemic mast cell disease is characterized by lymphadenopathy with or without peripheral blood eosinophilia. Patients with this rare disorder often lack cutaneous lesions, but frequently have mast cell infiltrates involving the bone marrow, gastrointestinal tract, liver, spleen, and lymph nodes. MCL is an extremely rare condition, and the diagnosis is established by demonstrating MCs in the peripheral blood and/or >20% MCs in a bone marrow aspirate smear. Most MCL patients do not have cutaneous lesions, but frequently experience fever, weight loss, abdominal pain, diarrhea, nausea, and vomiting. These patients also have detectable hepatosplenomegaly and lymphadenopathy resulting from extensive mast cell infiltration of these organs. Bone marrow biopsies from MCL patients demonstrate increased MCs, which are often spindle-shaped or morphologically atypical. Mast cell sarcomas and extracutaneous mastocytomas are extremely rare, with only a few isolated case reports.
Cutaneous mastocytosis (CM)
Mast cell sarcoma Extracutaneous mastocytoma |
| Major |
| Multifocal dense mast cell infiltrates (>15 MCs/aggregate) in the bone marrow or extracutaneous organs |
| Minor |
| 25% MCs are spindle-shaped or atypical in a bone marrow aspirate smear or tissue sections |
| c-KIT mutation at codon 816 in blood, bone marrow, or extracutaneous tissue |
| Expression of CD2 and/or CD25 by CD117 + MCs |
| Total serum tryptase persistently >20 ng/mL (in the absence of another nonmast cell hematologic disorder) |
Clinical Manifestations
Symptoms
Symptoms associated with mast cell disease are attributable in great part to the release of mast cell mediators, such as histamine, eicosanoids, and cytokines ( Table 42-3 ). These symptoms may range from pruritus and flushing to abdominal pain and diarrhea, to palpitations, dizziness, and syncope ( Table 42-4 ). In many instances the symptoms can be reduced or suppressed by antihistamines, suggesting mast cell-derived histamine as a cause. Many patients with CM or ISM have few, if any, symptoms, and may experience only intermittent bouts of pruritus. Of interest is the relative lack of pulmonary symptoms in SM patients. Complaints of fever, night sweats, malaise, weight loss, bone pain, epigastric distress, and problems with mentation (cognitive disorganization) often signal the presence of SM. Symptoms of mast cell disease can be exacerbated by exercise, heat, local trauma to skin lesions, as well as the ingestion of alcohol, narcotics, salicylates, and anticholinergic agents. Some systemic anesthetic agents (e.g., opiates) also may precipitate anaphylaxis.
| Preformed Mediators | Cytokines |
| Histamine | TNF-α |
| Heparin | IL-1 |
| IL-3 | |
| IL-4 | |
| Chemotactic factors for polymorphonuclear neutrophils and eosinophils Tryptase Chymase | IL-5 IL-6 IL-8 IL-9 IL-10 IL-13 IL-16 IL-18 SCF GM-CSF |
| Newly Formed Mediators | |
| PGD 2, | |
| LTC 4 , LTD 4 , LTE 4 | |
| PAF | |
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


