Abstract
Neurocutaneous genetic disorders, also called phakomatoses, manifest with various cutaneous and neurologic findings. Examples include neurofibromatosis, tuberous sclerosis, Sturge–Weber syndrome, and von Hippel–Lindau syndrome, the first three of which have striking cutaneous manifestations. Neurofibromatosis is an autosomal dominant disorder characterized by cutaneous neurofibromas, café-au-lait macules, and myriad systemic features with marked variability of expression. Dermatologic features of tuberous sclerosis complex can be easily recognizable and are present in >90% of patients with tuberous sclerosis complex. Sturge–Weber syndrome is a sporadic congenital condition characterized by a facial capillary malformation (port-wine stain) in association with leptomeningeal angiomatosis and glaucoma. Knowledge of the pathogenesis of these disorders had been static for many years. However, advances in gene identification have led to the discovery of genes responsible for some of these neurocutaneous disorders. Identification of the genes, their products, and roles in pathogenesis has led to significant clinical advances in treatment.
Keywords
Angiofibroma, Ataxia–telangiectasia, Neurocutaneous, Neurofibroma, Neurofibromatosis, Sturge–Weber syndrome, Tuberous sclerosis complex, Von Hippel–Lindau syndrome
- •
Neurofibromatosis is an autosomal dominant disorder characterized by cutaneous neurofibromas, café-au-lait macules, and myriad systemic features with marked variability of expression.
- •
Patients with neurofibromatosis are at risk for the development of a malignant peripheral nerve sheath tumor.
- •
Increasing or constant pain, change in consistency, or rapid growth of a nodule within an existing plexiform neurofibroma are concerning signs of malignant transformation.
- •
Tuberous sclerosis complex (TSC) is an autosomal dominant, multisystem disorder characterized by the development of hamartomas in multiple organ systems, including the brain, eyes, heart, lung, liver, kidneys, and skin.
- •
Dermatologic features of TSC can be easily recognizable and are present in >90% of patients with TSC.
- •
Knowledge of the functional relationship between TSC1/TSC2 and mTORC1 has led to important clinical advances in the use of mTOR inhibitors for the treatment of several clinical manifestations of TSC.
- •
Sturge–Weber syndrome (SWS) is a sporadic congenital condition characterized by a facial capillary malformation (port-wine stain [PWS]) in association with leptomeningeal angiomatosis and glaucoma.
- •
The neurologic features of SWS may be progressive and include seizures, focal neurologic impairment, and cognitive deficits.
- •
Ataxia–telangiectasia (AT) is an autosomal recessive disorder consisting of progressive cerebellar ataxia, ocular and cutaneous telangiectasia, and variable immune deficiency.
- •
Approximately 10–25% of patients with AT develop a malignancy, the majority of which are lymphoproliferative disorders.
Discussions of neurocutaneous disease are frequently limited to descriptions of the four “phakomatoses.” These conditions were grouped together because they all involved central nervous system (CNS) and retinal tumors (phakomas). The phakomatoses include neurofibromatosis, tuberous sclerosis complex, Sturge-Weber syndrome, and von Hippel–Lindau syndrome (VHL), the first three of which have striking cutaneous manifestations. Knowledge of the pathogenesis of these disorders had been static for many years. However, advances in gene identification have led to the discovery of the genes responsible for these neurocutaneous disorders. Identification of the genes, their products and role in pathogenesis has led to significant clinical advances in treatment. This discussion provides a broad overview of the most important neurocutaneous disorders as well as a variety of other conditions in which cutaneous and nervous system findings are shared.
Classification
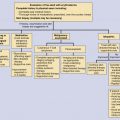
The relationship between the skin and the nervous system may be based on: (1) a developmental abnormality, frequently a result of a shared embryogenesis; or (2) the systemic effect on both organ systems of a metabolic disorder, infection, or immune response. The developmental and metabolic disorders are primarily of genetic origin, whereas the infectious and immune abnormalities represent the response of the skin and nervous system to a common insult. The neural crest is a transient embryonic structure that gives rise to dorsal root ganglion cells, Schwann cells, autonomic ganglion cells, as well as melanocytes. Abnormalities of the neural crest cells lead to a myriad of clinical findings. Unfortunately, the resulting disease entities can seldom be distilled to a pattern that can be totally explained by deductive reasoning. Disorders involving both the skin and the nervous system can be briefly categorized if one includes only the phakomatoses, but the list becomes very extensive if less common syndromes and systemic diseases involving both organ systems are included. The classifications in Table 40-1 include the classic neurocutaneous disorders and representative examples of various syndromes.
|
The term “phakomatosis” is derived from the Greek word “phakos” meaning “mother spot or mole.” Although originally used to describe the retinal lesions of tuberous sclerosis, it has come to refer to a group of disorders including: (1) tuberous sclerosis complex; (2) neurofibromatosis; (3) Sturge–Weber disease; and (4) VHL syndrome. These disorders, as well as other vascular abnormalities, will be discussed in detail. The remainder of the developmental disorders represents disorders of epidermal cells and their appendages that share an ectodermal origin with the nervous system. The syndromes mentioned in Table 40-1 are rare but represent examples of probable neural crest abnormalities as well as poorly understood but well-documented disorders involving both the skin and the nervous system.
Neurofibromatosis (von Recklinghausen Disease)
Neurofibromas may occur in several clinical settings. Sporadic solitary cutaneous tumors can arise in adulthood and are not associated with café-au-lait spots. There are three major clinically and genetically distinguishable forms of neurofibromatosis: neurofibromatosis type 1, neurofibromatosis type 2, and schwannomatosis. Classic neurofibromatosis (type 1 or NF1—OMIM #162200) as described by von Recklinghausen is characterized by multiple café-au-lait macules, cutaneous neurofibromas, and myriad systemic involvement with marked variability of expression. Acoustic neurofibromatosis (type 2 or NF2—OMIM #101000) presents with bilateral acoustic neuromas as well as café-au-lait macules and cutaneous neurofibromas. The gene for NF2 is localized on chromosome 22 and encodes a negative growth regulator called MERLIN. Schwannomatosis (neurilemmomatosis—OMIM #162091) is characterized by multiple noncutaneous schwannomas in the absence of acoustic tumors. Mutations in the tumor suppressor genes SMARCB1 and LZTR1 are thought to be responsible for the majority of cases. Segmental (dermatomal) neurofibromatosis is a syndrome where café-au-lait macules, cutaneous neurofibromas, and sometimes visceral neurofibromas are limited to a sharply defined unilateral body segment. Watson’s syndrome, a variant of NF1, has multiple café-au-lait macules, short stature, and pulmonary valvular stenosis, but only a small number of neurofibromas. This discussion will be limited to NF1 and the term “neurofibromatosis” is used to refer to only that disorder.
Pathogenesis
Neurofibromatosis is an autosomal dominant disorder affecting approximately 1 in 3000 live births. De novo mutations represent around 50% of patients with neurofibromatosis. Penetrance approaches 100% by age 20, but the expressivity is highly variable. With an estimated mutation rate of 1 in 10,000 per gamete per generation, the NF1 gene has one of the highest mutation rates of any genetic disorder. Currently over 1000 pathogenic variants in NF1 have been identified. About 90% of new mutations occur on the paternally derived chromosome with the majority causing truncation of the gene product.
The NF1 gene has been localized to chromosome 17q11.2. The basic defect lies in the abnormal expression of a tumor suppressor gene called neurofibromin. This gene product has functional and structural homology with a guanosine triphosphatase-activation protein (GTPase) that downregulates p21 ras proto-oncogene activity. In its active (GTP) state, the p21 ras protein binds guanine nucleotides with high affinity, acting as a signal transducer for cellular proliferation. Neurofibromin switches off this signaling by hydrolyzing GTP to guanosine diphosphate ( Fig. 40-1 ). A defect in neurofibromin would result in a constitutively active p21 ras protein leading to cell growth and possible tumor formation. Interestingly, neurofibromin is found in the skin, brain, spleen, liver, and muscle, not just in the neural crest cells. The abnormal proliferation seen in neurofibromatosis is focused on the neural crest for reasons that are not yet clear at the molecular level.
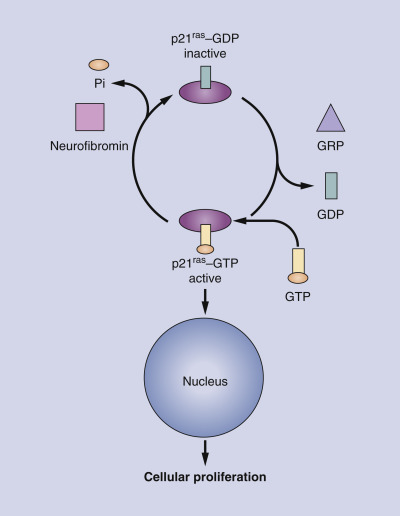
It is generally believed that a somatic “second hit” mutation with loss of heterozygosity results in the progression of malignant tumors and possibly in the formation of benign neurofibromas. However, given the wide clinical phenotypes found even among close relatives, a defect in the neurofibromin protein cannot in itself explain this variability. Other studies point to evidence that modifying genes (genes that are unlinked to the NF1 gene, but play a phenotypical role in neurofibromatosis) may be behind these differences. Clinical outcomes would depend on where the second mutation at another gene occurs. Finally, recent research has shown that the environment, especially trauma, may lead to the formation of benign neurofibromas. Since neurofibromin is involved in the healing process, defective neurofibromin in conjunction with a traumatic event may lead to abnormal cellular proliferation and tumor formation.
Clinical Manifestations
Café-au-Lait Macules
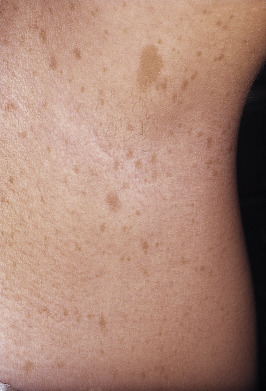
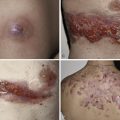
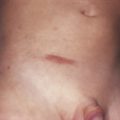
Present in virtually all patients, these hyperpigmented macules are the earliest clinical features found in neurofibromatosis ( Table 40-2 ). Up to 15% of the normal population have one to three café-au-lait spots. The current criterion for the disease is the presence of six or more café-au-lait macules larger than 1.5 cm for adults and 0.5 cm for prepubertal individuals ( Fig. 40-2 ). Café-au-lait spots are usually present at birth, or shortly thereafter, and progressively increase in number and size. The macular hyperpigmentation is usually homogeneous with sharply defined edges. Distribution is random, sparing the scalp, palms, and soles They tend to darken in response to sunlight and will fade with time and become less noticeable.
| Café-au-lait macules |
| Axillary freckling |
| Lisch nodules |
| Neurofibromas |
Skin Fold Freckling
The freckle-like pathognomonic lesions (Crowe’s sign) ( Fig. 40-2 ) generally occur between 3 and 5 years of age in the axillae and/or groin. Around 90% of adults have the freckling. These freckles are typically small, distinguishing them from café-au-lait macules. Other involved areas include the area above the eyelids, around the neck, and submammary region in females.
Neurofibromas
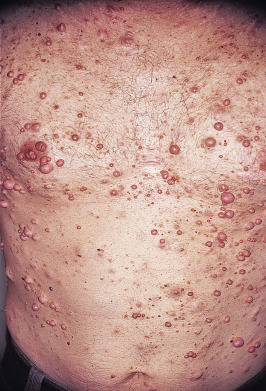
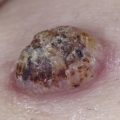
Neurofibromas may be subdivided according to their appearance and location into the following groups: cutaneous, nodular (subcutaneous), and plexiform. All are composed of various combinations of Schwann cells, fibroblasts, perineural cells, vascular elements, and mast cells. Cutaneous neurofibromas are rarely present in infancy; more often they begin to appear in late childhood and adolescence and gradually increase in size and number. Clinically, they are soft, fleshy, sessile, or pedunculated tumors that can be invaginated (buttonhole sign) ( Fig. 40-3 ). Subcutaneous, or nodular, neurofibromas tend to become apparent in late childhood or early adulthood. Subcutaneous lesions can be noted on palpation of the skin and may present with tenderness or tingling distributed along the affected nerve. Both pregnancy and puberty are known to increase the number and size of neurofibromas. Plexiform neurofibromas may be superficial, deep, or a combination of both. These tumors arise from multiple nerve fascicles, tend to grow along the length of a nerve, and can feel like a “bag of worms.” They can extend into surrounding structures, including the skin, fascia, muscle, bone, and internal organs. They can be painful and affect the growth and function of the affected area.
Patients with neurofibromatosis are at risk for the development of a malignant peripheral nerve sheath tumor (MPNST). Patients with NF1 have a 10% lifetime risk of developing this highly aggressive spindle cell tumor. MPNSTs typically arise from plexiform neurofibromas. Significant and constant pain, change in consistency, or rapid growth of a nodule within an existing plexiform neurofibroma are concerning signs of malignant transformation.
Lisch Nodules
These pigmented iris hamartomas are present in >90% of adults with neurofibromatosis. They are asymptomatic and are not correlated with other manifestations or with severity of the disease. Clinically they appear as randomly distributed cream-colored to brownish nodules on the iris. While most are easy to visualize, a slit-lamp evaluation by an experienced ophthalmologist is often needed to rule out the presence of single “salt grain” lesions. Histologically, they represent melanocytic hamartomas.
Central Nervous System Involvement
CNS tumors develop in 3% to 10% of patients and include benign neoplasms such as optic nerve gliomas, acoustic neuromas, neurolemmomas, meningiomas, ependymomas, astrocytomas, and neurofibromas. Such growths present clinically with signs and symptoms of CNS mass lesions. Spinal tumors often present with localizing peripheral signs. Optic pathway glioma (OPG), the most common of these tumors, involves some combination of the optic nerves, chiasm, or optic tract. They may result in papilledema, retrobulbar neuritis, and eventually optic atrophy. Some may manifest as precocious puberty due to hypothalamic encroachment. Most OPGs develop within the first 6 years of life and the majority of cases run a benign course and do not require intervention. For those whose tumors do progress, chemotherapy is the treatment of choice. Malignant tumors, most commonly low-grade astrocytomas, may also occur. They affect the brainstem and may present with symptoms of mass effect such as cranial neuropathies and hydrocephaly. However, these lesions tend to have a less aggressive course when compared to pontine tumors that are not associated with neurofibromatosis.
A more recent issue is the presence of high-intensity signals on T2 magnetic resonance imaging (MRI). These NF-associated bright spots (also called unidentified bright objects) are often found within the basal ganglia, cerebellum, and brainstem in up to 70% to 80% of patients with neurofibromatosis. What these images represent remains unclear. Current speculation includes harmatomas, dysplasia, demyelination, vacuolar changes, or low-grade tumors. The clinical significance of these findings is also in dispute. Some studies have shown that the presence of bright spots is associated with lower IQ scores, but this conclusion has remained controversial. Some practitioners believe that the findings of bright spots should be included as a criterion for the diagnosis of neurofibromatosis and may be diagnostic in 30% of the cases of children who do not meet the current criteria. However, consideration must be taken in regard to children who may need anesthesia in order to have an MRI performed.
Learning disabilities occur in 50% to 75% of patients. Frank intellectual disability is seen in about 5% of patients. The learning problems associated with neurofibromatosis persist into adulthood. There is a higher incidence of attention-deficit disorder, autism spectrum disorders, behavioral abnormalities, and psychosocial issues. Headaches occur with increased frequency even in the absence of CNS tumors. Mild speech impediments are present in 30% to 40% of cases, and cerebrovascular compromise as a result of involvement of cerebral arteries with neurofibromas does occur. Major and minor motor seizures occur in less than 5% of cases in the absence of identifiable mass lesions or cerebral vascular involvement. An additional 26% of patients will have abnormal or borderline findings on electroencephalogram.
Macrocephaly is present in at least 27% of patients. This manifestation is more common after 6 years of age. There is no correlation between macrocephaly and impaired intellectual performance, seizures, or electroencephalographic abnormalities.
Musculoskeletal Disorders
Sphenoid wing dysplasia is a distinctive osteopathy of neurofibromatosis. It presents as a unilateral defect affecting the orbital plate and frontal bone. Dysplasia of a long bone occurs in nearly 14% of patients with neurofibromatosis and usually presents in the first year of life. The most commonly affected bone is the tibia, which will bow in an anterolateral direction. Repeated fracture and failure to heal can result in a pseudoarthrosis, which is a false joint. Scoliosis has been reported to occur in up to 10% of patients with neurofibromatosis and usually becomes apparent by age 10 years. Dystrophic scoliosis is a rapidly progressive form with the potential to cause a variety of neurologic complications. Radiographic bone abnormalities may also result from pressure by intraosseous or paraosseous neurofibromas. No consensus on treatment is available and severe cases have led to amputation. Short stature is also another feature more prominent in individuals with neurofibromatosis. Growth curves comparing nonaffected and affected children show similar growth profiles until preadolescence at which point the growth rate for children with neurofibromatosis decreases significantly. In addition, they are at increased risk of osteoporosis and osteopenia later in life.
Vascular Disorders
Hypertension is more prevalent in patients with NF1 and can develop at any age. The hypertension is usually essential, but may occur secondary to renal artery stenosis, coarctation of the aorta, or pheochromocytoma. Vasculopathy may involve arteries of the heart and brain. Congenital heart defects and valvar pulmonic stenosis are more common in individuals with neurofibromatosis.
Gastrointestinal Disorders
Visceral tumors arise from intra-abdominal neural tissue and include neurofibromas, leiomyomas, and miscellaneous tumors. The most common complications are obstruction, intussusception, and hemorrhage caused by erosions or necrosis of pedunculated tumors. Persistent constipation occurs in 10% of cases and is due to disorganization of the tunica muscularis and Auerbach’s plexus of the colon.
Endocrine Disorders
Aberrant endocrine function is not a regular finding in neurofibromatosis and although frequently mentioned is probably present in less than 1% of cases. Pheochromocytoma, often benign and unilateral, is the most common endocrine abnormality. It predominantly produces norepinephrine. Medullary carcinoma of the thyroid and hyperparathyroidism are even less common. In addition, it has been reported that optic gliomas in children with hypothalamic involvement may present with either premature or delayed puberty.
Miscellaneous Disorders
Miscellaneous malignancies, including Wilms’ tumor, rhabdomyosarcoma, and leukemia, and a variety of tumors of neural crest origin, including malignant melanomas, are more common in patients with neurofibromatosis than in the population at large. An association with juvenile xanthogranuloma and juvenile myelomonocytic leukemia has been found in children with neurofibromatosis. It is felt that the loss of the tumor suppression function of the NF1 gene predisposes those with neurofibromatosis to myeloid disorders.
Differential Diagnosis
The diagnostic criteria outlined by the US National Institutes of Health are based upon specific clinical features of NF1. Two or more of the following criteria are needed:
- 1.
Six or more café-au-lait spots greater than 1.5 cm in adults and 0.5 cm in prepubertal children.
- 2.
Two or more neurofibromas of any type or one plexiform neurofibroma.
- 3.
Axillary or inguinal freckling.
- 4.
Optic glioma.
- 5.
Two or more Lisch nodules.
- 6.
A distinctive bony lesion, such as sphenoid dysplasia or thickening of a long bone cortex with or without pseudoarthrosis.
- 7.
A first-degree relative with NF1.
These diagnostic criteria are both highly sensitive and specific for adults with neurofibromatosis. They are less sensitive in children, especially those under 8 years of age, because some of the criteria do not manifest at a young age. Other minor features that may assist in the diagnosis include macrocephaly, hypertelorism, short stature, and thorax abnormalities.
Legius syndrome (OMIM #611431), an autosomal dominant NF1-like disorder, is a genetically distinct disorder with a similar phenotype to NF1. It results from germline loss-of-function mutations in SPRED1. The SPRED1 gene encodes a negative regulator of the RAS-MAPK pathway, similar to neurofibromin. This disorder is characterized by multiple CALMs, axillary freckling, and macrocephaly. These individuals do not have neurofibromas, Lisch nodules, or NF1 gene abnormalities.
Patient Evaluation
Given the myriad of clinical features in neurofibromatosis, the utility of extensive screening tests remains controversial. In general, clinical evaluation tends to be more helpful to detect complications than are screening investigations in asymptomatic patients. Annual visits should include a thorough physical exam, a formal ophthalmologic exam, assessment for precocious puberty, developmental assessment, review of school performance, and monitoring of plexiform neurofibromas.
Genetic Testing
With the current methodologies available approximately 85% to 95% of the currently known mutations can be identified. In familial cases of neurofibromatosis, indirect linkage studies can be performed. Genes are said to be linked when markers close to the NF1 locus segregate together during recombination. Through the extensive evaluation of multiple family members, analysis of the markers associated with the abnormal NF1 gene can be identified and used for genetic counseling. In addition, in utero diagnosis studies are currently available. However, genetic testing in neurofibromatosis remains problematic. The large variability in phenotypic expression of neurofibromatosis does not correlate with specific genetic mutations. Positive genetic results do not predict the presence, age of onset, or the severity of disease. There are two exceptions. Patients with whole gene deletions, which occur in 4% to 5% of patients with NF1, present with a large tumor burden, more severe cognitive impairment, large hands and feet, dysmorphic facial features, and have a higher lifetime risk of developing MPNSTs. A three-base-pair in-frame deletion in exon 17 of the NF1 gene is the second exception. Patients with this genetic mutation have an absence of cutaneous neurofibromas and appear to have a lower incidence of serious complications.
Negative genetic analysis does not exclude the disease. As a result the demand for prenatal testing remains limited. Genetic testing may offer benefit as a diagnostic tool in suspected patients who do not meet criteria. However, as the cost and the prognostic capabilities of these tests improve, genetic testing may play a more significant role in the future.
Treatment
Neurofibromatosis is a multisystem disorder requiring management by multiple disciplines. Current management focuses on genetic counseling and symptomatic treatment of specific complications. Patients with NF1 have an estimated lifespan that is 15 years lower than the general population with malignant degeneration being the major cause of early death. There is no overall treatment for neurofibromatosis. Individual manifestations and complications are treated as they arise. Regular follow-up and surgical removal of rapidly enlarging lesions should be undertaken. In addition, removal of disfiguring or functionally compromising lesions is recommended. The removal of subcutaneous neurofibromas may result in neurologic deficit and should be performed by a skilled surgeon. Various medical treatments for plexiform neurofibromas are being evaluated in ongoing clinical trials.
The cosmetic, medical, behavioral, and social features of this disease may diminish the quality of life in patients with neurofibromatosis. Awareness of the various complications and methods of coping with them are essential to decrease long-term morbidity rate.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


