Abstract
Dermatomyositis is a condition that combines an inflammatory myopathy with a characteristic cutaneous disease. Its pathogenesis is only partially understood, but immune-mediated muscle damage is believed to be important as a pathogenetic mechanism. Dermatomyositis appears to be characterized by an increased frequency of internal malignancy. The characteristic and possibly pathognomonic cutaneous features of dermatomyositis are the heliotrope eruption and Gottron’s papules. Several other cutaneous features that occur in patients who have dermatomyositis are characteristic of the disease but are not pathognomonic, including mid-facial erythema, poikiloderma in a photosensitive distribution on the chest (“V-neck” sign) or back (shawl sign), a violaceous erythema on the extensor surfaces, alopecia with or without scaly poikilodermatous changes, a scaly poikilodermatous rash on the lateral thighs (Holster sign), and periungual and cuticular changes. Dermatomyositis is a multisystem disorder. The overall therapeutic plan is determined primarily by the presence or absence of myositis or other internal organ involvement. The mainstay of therapy for myositis is the use of systemic corticosteroids. Therapy for cutaneous disease in patients with dermatomyositis is often difficult because, even though the myositis may respond to treatment with corticosteroids and/or immunosuppressants, the cutaneous lesions often persist.
Keywords
Antisynthetase syndrome, Amyopathic dermatomyositis, Calcinosis cutis, Gottron’s papules, Heliotrope eruption, Idiopathic inflammatory myopathy, Myositis-specific autoantibodies, Poikiloderma
- •
Classic dermatomyositis involves a proximal inflammatory myopathy with a characteristic cutaneous eruption. In patients with clinically amyopathic dermatomyositis, skin disease is the prominent feature.
- •
Pathognomonic cutaneous findings of dermatomyositis include the heliotrope eruption and Gottron’s papules. In dermatomyositis, cutaneous disease is photoexacerbated.
- •
All adult patients with dermatomyositis require screening for malignancy and pulmonary disease, regardless of whether they have muscle involvement.
- •
Juvenile dermatomyositis is not associated with an increased risk of cancer, but does have an increased association with calcinosis cutis and vasculitis.
- •
In dermatomyositis, treatment for and therapeutic response of muscle and skin disease often differ. Cutaneous dermatomyositis is classically challenging to treat and negatively impacts patient quality of life.
Definition and Classification
Dermatomyositis is a condition that combines an inflammatory myopathy with a characteristic cutaneous disease. A closely related disease, polymyositis, has all the clinical features of the muscular disease of dermatomyositis but lacks the characteristic cutaneous findings. A third idiopathic inflammatory myopathy, inclusion body myositis, also lacks cutaneous disease, but has a unique pattern of weakness with prominent involvement of the wrist and finger flexors and quadriceps. The pathogenesis of these disorders is only partially understood, but immune-mediated muscle damage is believed to be important as a pathogenetic mechanism. Dermatomyositis appears to be characterized by an increased frequency of internal malignancy, whereas the association of polymyositis with malignancy is less well resolved. There is a female to male preponderance of approximately 2:1, with a peak incidence in the fifth and sixth decades. A juvenile form of dermatomyositis also exists, which is not associated with an increased risk of malignancy, but does have an increased incidence of calcinosis cutis and vasculitis. Because both dermatomyositis and polymyositis are associated with morbidity and occasional deaths, a prompt and aggressive approach to therapy is indicated.
Bohan and Peter first suggested the use of five criteria to diagnose dermatomyositis. These include (1) proximal symmetric muscle weakness that progresses over a period of weeks to months; (2) elevated serum levels of muscle-derived enzymes; (3) an abnormal electromyogram; (4) an abnormal muscle biopsy; and (5) the presence of cutaneous disease compatible with dermatomyositis. These criteria are useful for patient evaluation, but it is not necessary to perform all of the muscle testing in patients with characteristic skin disease, particularly those who have proximal muscle weakness and elevated muscle-derived enzymes.
The inflammatory myopathies may be subclassified into eight groups. The following system of classification has been useful in differentiating groups of patients with regard to their prognosis, potential for an associated process, and potential to respond to various therapies: (1) dermatomyositis; (2) polymyositis; (3) myositis in association with malignancy; (4) juvenile dermatomyositis (most often diagnosed before age 16); (5) myositis in association with another connective tissue disease; (6) inclusion body myositis; (7) dermatomyositis sine myositis (amyopathic dermatomyositis); and (8) drug-induced disease.
Given that the incidence and prevalence of amyopathic dermatomyositis appear to be increasing, Sontheimer has proposed a revised classification system that better recognizes the cutaneous manifestations of the inflammatory myopathies ( Table 2-1 ).
| Dermatomyositis (DM) |
|
| Polymyositis |
| Inclusion body myositis |
Pathogenesis
The pathogenesis of the idiopathic inflammatory myopathies is not well understood. The pathogenetic mechanisms involved in the muscular disease are better understood than are those involved in the induction of cutaneous disease. Many agents and events have been associated with the appearance of dermatomyositis, including various infections (particularly viral or parasitic infections), vaccination, neoplasms, drug-induced disease, various types of stress, and trauma. In addition, dermatomyositis and polymyositis have been linked with various diseases associated with immunologic phenomena. The demonstration of the Jo-1 antibody in patients with myositis further supports a viral etiology because the antigen for the Jo-1 antibody has characteristics similar to those of viral and muscle proteins. Patients with active dermatomyositis or polymyositis have been demonstrated to have upregulation of type I interferon-α/β-inducible genes in blood samples, and the level of type I interferons has been shown to correlate with disease activity. Despite having this upregulation of type I interferons in common, it is thought that the immunopathogenesis of dermatomyositis and polymyositis is different. In polymyositis, clonally expanded autoreactive CD8-positive T cells invade myocytes expressing major histocompatibility complex (MHC) class I antigens and cause necrosis via the perforin pathway. In dermatomyositis, autoantigens activate a humoral immune process in which complement is deposited in capillaries, causing capillary necrosis and ischemia. For both diseases, a genetic predisposition has also been suggested. Thus, under appropriate circumstances in an immunogenetically predisposed individual, an infection, drug, trauma, or neoplasm may be able to initiate an inflammatory reaction in the muscle and skin. Through a complex set of reactions involving immunologic phenomena, muscle damage and cutaneous disease may occur.
Manifestations
Cutaneous Manifestations
The characteristic and possibly pathognomonic cutaneous features of dermatomyositis are the heliotrope eruption and Gottron’s papules. Several other cutaneous features that occur in patients with dermatomyositis are characteristic of the disease but are not pathognomonic. These include midfacial erythema that involves the nasolabial folds (as opposed to the malar erythema in lupus erythematosus, which spares the nasolabial folds), poikiloderma in a photosensitive distribution on the chest (“V-neck” sign) or back (shawl sign), a violaceous erythema on the extensor surfaces, alopecia with or without scaly poikilodermatous changes, a scaly poikilodermatous rash on the lateral thighs (Holster sign), and periungual and cuticular changes. The cutaneous disease in dermatomyositis is photodistributed and often photoaggravated. In addition, pruritus may be a prominent feature in dermatomyositis, and may be helpful in clinically distinguishing this entity from lupus erythematosus. Quality-of-life impairment in dermatomyositis is greater than in other skin diseases, including both psoriasis and atopic dermatitis, based on its cutaneous manifestations alone. Notably, the cutaneous manifestations of dermatomyositis can follow the course of the myositis or can be discordant from the muscle disease activity. Reactivation of any skin manifestation of dermatomyositis in a patient otherwise considered to be in remission may signify a relapse of the myositis. Most often, however, the activity of the muscle disease is not reflected by that of the cutaneous disease.
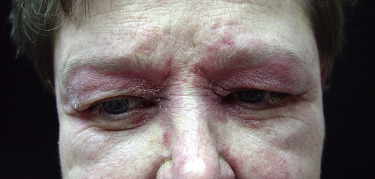
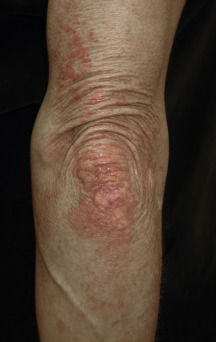
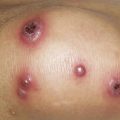
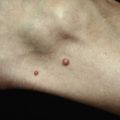
The heliotrope eruption consists of a dusky erythematous to violaceous cutaneous eruption with or without edema involving the periorbital skin in a symmetric distribution ( Fig. 2-1 ). Often only the upper lid is involved. Sometimes this sign is subtle and may involve only faint erythema along the eyelid margin. Gottron’s papules are found over bony prominences, particularly the metacarpophalangeal joints, the proximal interphalangeal joints, and/or the distal interphalangeal joints. They may also be found over bony prominences such as the elbows, knees, and feet. The lesions consist of slightly elevated erythematous to violaceous papules and plaques ( Figs. 2-2 and 2-3 ); they often contain telangiectases, and there may be hyper- and/or hypopigmentation. These lesions can be clinically confused with those of lupus erythematosus or, at times, with those of papulosquamous disorders such as psoriasis or lichen planus. In instances in which differentiation is difficult, a biopsy is warranted; however, the features of dermatomyositis are indistinguishable from those of cutaneous lupus erythematosus. Gottron’s sign typically refers to violaceous erythema over the areas where Gottron’s papules tend to occur.
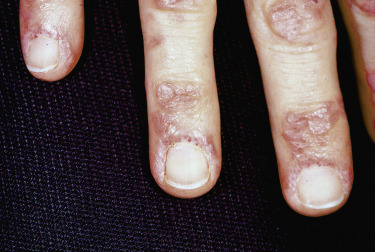
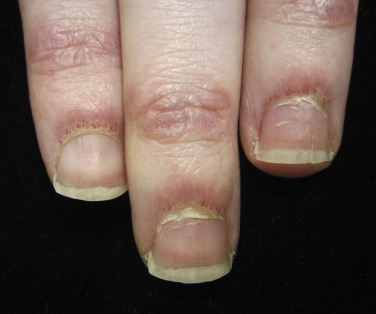
Nailfold changes consist of periungual telangiectases (consisting of both dilated capillary loops and capillary dropout), cuticular hypertrophy, and small hemorrhagic infarcts within this hypertrophic area ( Fig. 2-4 ). The periungual telangiectases may be clinically apparent or may be appreciated only by capillary microscopy. Clinically, they may closely resemble those seen in other connective tissue diseases. The cuticular overgrowth may be similar to that seen in scleroderma.
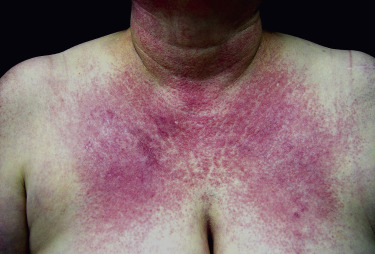
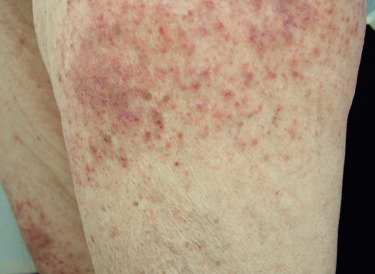
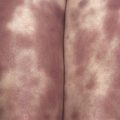
Poikiloderma can occur on any photodistributed site, most classically the upper chest and neck (“V-neck” sign) and the upper back (shawl sign) ( Fig. 2-5 ), but also on the extensor surfaces of the arms and within Gottron’s papules on the dorsal hands. This photosensitive poikilodermatous eruption must be differentiated from lupus erythematosus and from other diseases that cause poikilodermatous skin changes. In addition, poikilodermatous changes may also occur on the lateral thighs, known as the “holster sign” ( Fig. 2-6 ).
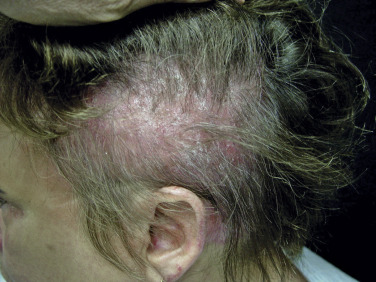
Scalp involvement in dermatomyositis is relatively common. Mild to moderate nonscarring alopecia ( Fig. 2-7 ) can occur in some patients and often follows a flare of the disease. In addition, the scalp is often diffusely affected by scaling and erythema with poikilodermatous features, which can result in intense scalp pruritus. The scalp pruritus is often severe, and the symptoms often are greater than one might appreciate from the clinical findings. Clinical distinction from seborrheic dermatitis or psoriasis may be challenging, but histopathologic evaluation is helpful.
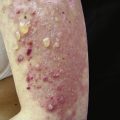
Less common cutaneous findings include an exfoliative erythroderma as well as vesiculobullous, erosive, and ulcerative lesions. Patients with myositis can also develop the lesions of other collagen–vascular diseases. The presence of these types of lesions allows physicians to classify the patients into an overlap category. In general, sclerodermatous skin changes have been the most frequently seen in patients with overlap syndrome. However, cutaneous vasculitis, discoid lupus erythematosus, and rheumatoid nodules have also been known to occur in patients with dermatomyositis.
Skin biopsy may aid in differentiating dermatomyositis from other papulosquamous or eczematous diseases, but cannot be used to reliably distinguish dermatomyositis from lupus erythematosus. In addition, direct immunofluorescence microscopy is usually not helpful in differentiating lupus erythematosus and dermatomyositis. Classically, skin biopsy in dermatomyositis demonstrates a vacuolar interface dermatitis with mucin deposition in the dermis. Cutaneous lesions of dermatomyositis that do not demonstrate the interface change classically observed with the pathognomonic and characteristic skin lesions include mechanic’s hands (hyperkeratosis of the lateral fingers and palms), panniculitis, cutaneous vasculitis, urticaria, a flagellate erythema, and follicular hyperkeratosis.
Although cutaneous lesions precede muscle disease in 30% to 56% of patients with classic dermatomyositis, myositis follows within 3 to 6 months in most cases. In a significant portion of patients, the myositis resolves with therapy, whereas the cutaneous disease persists and becomes the most salient feature of disease (postmyopathic dermatomyositis). Amyopathic dermatomyositis is provisionally diagnosed when typical cutaneous disease is present for at least 6 months without clinical weakness, with repeatedly normal serum muscle enzyme levels, and in patients who have not been treated with systemic corticosteroids or immunomodulatory agents for more than a few months. The diagnosis of amyopathic dermatomyositis is considered confirmed after skin disease has been present for 2 years in the absence of muscle involvement. Predictive factors for progression to classic dermatomyositis with muscle involvement have not yet been identified in this subset of patients.
Muscle Disease
Clinical and laboratory abnormalities that suggest muscle disease are characteristic features of polymyositis and dermatomyositis. Even in patients who have only cutaneous disease at presentation, myositis follows in the majority of patients. Myositis precedes the cutaneous findings in less than 10% of patients. The myositis occurring in dermatomyositis is indistinguishable from that occurring in polymyositis as assessed by clinical and laboratory features. Also, when considered alone, the individual features of myositis are not diagnostic of dermatomyositis or polymyositis; rather, the diagnosis is one of exclusion.
Clinically, the myopathy affects mainly the proximal muscle groups of the shoulder and pelvic girdle. In severe, progressive disease all muscles may become involved. The disease is usually symmetric. The initial complaints include weakness, fatigue, an inability to climb stairs, an inability to raise the arms for actions such as hair grooming or shaving, an inability to rise from a squatting or sitting position, or a combination of these features. The progression of disease is variable, but usually occurs over a period of weeks to months. Muscle aching is a common subjective complaint, but frank tenderness on palpation is variable. An inability to swallow and symptoms of aspiration may reflect the involvement of striated muscle of the pharynx or upper esophagus. Dysphagia often signifies a rapidly progressive course and may be associated with a poor prognosis.
Muscle enzyme levels are frequently elevated in patients with inflammatory myopathy. The enzymes that are commonly elevated are creatine kinase, aldolase, lactic dehydrogenase, and/or serum transaminases. In the vast majority of patients, creatine kinase is the most practical test available for measuring the activity of muscle disease. Other potential abnormalities include disturbances of electrical action on electromyography (EMG), histopathologic changes (muscle biopsy classically demonstrates type II fiber atrophy, necrosis, regeneration, a centralization of the nuclei, and a lymphocytic infiltrate in a perifascicular and/or perivascular region), and/or abnormalities on magnetic resonance imaging (MRI) or ultrasound. The use of MRI may improve yield when performed prior to muscle biopsy or may demonstrate clinically inapparent inflammation. In children, levels of factor VIII-related antigen or neopterin may predict a more severe dermatomyositis variant with vasculopathy.
Systemic Features
Dermatomyositis and polymyositis are multisystem disorders. This is reflected by the high frequency of other clinical features in patients with these diseases.
Arthralgias and/or arthritis may be present in up to one-quarter of patients with inflammatory myopathy. This percentage rises in patients with overlap syndromes. The usual picture is one of generalized arthralgias accompanied by morning stiffness. The small joints of the hands, wrists, and ankles may be involved with symmetric nondeforming arthritis. Patients with arthritis may have a lower frequency of malignancy than those who do not have arthritis.
Esophageal disease as manifested by dysphagia is estimated to be present in 15% to 50% of patients with inflammatory myopathy. The dysphagia can be of two types: proximal or distal. Proximal dysphagia is caused by the involvement of striated muscle in the pharynx or proximal esophagus. This involvement correlates well with the severity of the muscle disease and is corticosteroid responsive. Distal dysphagia is related to the involvement of nonstriated muscle and appears to be more frequent in patients who have overlap syndromes. Distal dysphagia may also be accompanied by symptoms of reflux esophagitis. In general, dysphagia portends a poor prognosis and is often associated with pulmonary involvement.
Pulmonary disease occurs in approximately 15% to 30% of patients with dermatomyositis and polymyositis. It can be characterized by a primary diffuse interstitial fibrosis that may be manifested radiologically, or by abnormalities seen on pulmonary function testing. Pulmonary disease may also occur as a direct complication of the muscular disease, such as hypoventilation or aspiration in patients with dysphagia, or it may be a result of treatment, such as with opportunistic infections or drug-induced hypersensitivity pneumonitis. It is important to note that patients with amyopathic dermatomyositis may have aggressive lung disease even in the absence of myositis. Overall, pulmonary complications have been associated with a poor prognosis. Data have suggested that patients with myositis who have Jo-1 antibodies are at a greater risk for pulmonary involvement. In fact, 70% of patients with Jo-1 antibodies have interstitial lung disease. The antisynthetase syndrome is the constellation of interstitial lung disease, myositis, polyarthritis, Raynaud’s phenomenon, fever, and mechanic’s hands in a patient with antitransfer RNA autoantibodies.
Cardiac disease may also occur in patients with inflammatory myopathy, as manifested by myocarditis or pericarditis. Pericarditis appears to be more common in patients with overlapping features of other connective tissue diseases. Myocarditis can result in conduction defects, arrhythmias, or, when severe, congestive heart failure.
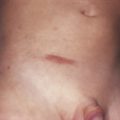
Calcinosis of the skin or muscle is unusual in adults but may occur in up to 40% of children with dermatomyositis. Calcinosis cutis is manifested by firm, yellow-white, or skin-colored nodules, often over bony prominences. Occasionally, these nodules can extrude through the surface of the skin, in which case secondary infections may occur. Calcification of the muscles is often asymptomatic and may be seen only on radiologic examination. In severe forms, the calcinosis can cause loss of function, and, rarely, bone formation is possible.
Pregnancy has been shown to have an effect on the inflammatory myopathy. In addition, the inflammatory myopathy may produce profound effects on the neonate and/or the mother. Studies suggest that dermatomyositis and/or polymyositis may be activated during pregnancy, or that the initial manifestations may be appreciated during pregnancy. In addition, in a large group of women with multiple pregnancies, premature delivery, spontaneous abortions, perinatal deaths, and fetal loss were more common in patients with active myositis.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


