- •
Sarcoidosis is a multisystem granulomatous disease of unknown etiology that affects patients of all ages and ethnic groups.
- •
Thorough multisystem evaluation of all patients is essential, as sarcoidal inflammation may be clinically quiescent while causing significant damage.
- •
Cutaneous sarcoidosis is characterized by a protean array of lesion morphologies, but generally occurs as violaceous infiltrated papules clustered on the face, particularly around the nose.
- •
Sarcoidosis may spontaneously resolve in up to two-thirds of patients during the first few years of disease; patients with extensive skin involvement are more likely to have a chronic course.
- •
Mortality occurs in 5% of patients, generally those with severe pulmonary involvement, cardiac involvement, or neurosarcoidosis.
- •
Treatment options range from topical and intralesional steroids, to antimalarials or anti-inflammatory antibiotics, to methotrexate, to TNF-α inhibitors.
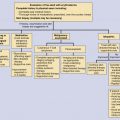
Sarcoidosis is a multisystem disorder of unknown cause characterized by the accumulation of T-cell lymphocytes, monocytes, and epithelioid macrophages from an unknown antigen(s) that induce the formation of noncaseating granulomas, leading to abnormal tissue and organ function. Sarcoidosis almost always involves the lungs; the skin, eyes, and lymph nodes are frequently involved; however, the disease can involve any organ system in the body. While there are suggestive laboratory and radiographic findings supportive of a diagnosis, there exists no gold-standard confirmatory diagnostic test. The disease is characterized by the presence of granulomatous inflammation in more than one organ. When this histopathologic change is present in tissue samples from multiple organ systems, and when appropriate tests exclude other causes of sarcoidal granulomas, the diagnosis of sarcoidosis can be confirmed.
The course of sarcoidosis is highly variable. It ranges from an acute self-healing process, to a chronic disease that primarily affects the skin, to a debilitating systemic disease that can result in blindness, progressive respiratory insufficiency, and sometimes death. Although associated with significant morbidity, sarcoidosis is rarely a primary cause of death, with fewer than 5% of patients dying of their disease, generally due to severe lung, neurologic, or cardiac involvement.
Cause and Pathogenesis
The cause of sarcoidosis is unknown. The current understanding entails a genetically susceptible host, who is then exposed to an environmental trigger, which sets off the host immune response leading to granuloma formation and the clinical disease phenotype. Over the years many causes have been postulated, including viruses (hepatitis C, HIV, HHV-8), bacteria ( Mycobacterium , Propionibacterium , Borrelia , Rickettsia , cell-wall-deficient bacteria), fungi (cryptococcus), an autoantigen or misfolded protein (amyloid A), and foreign materials (pine pollen, beryllium, talc, silica). Coexistent “autoimmune” diseases, such as autoimmune thyroiditis, alopecia areata, and vitiligo, have been reported with varying frequencies in small studies, and patients with various collagen–vascular diseases (scleroderma, lupus, or dermatomyositis) may be more prone to develop sarcoidosis.
There is no single gene that explains sarcoidosis; rather, the disease is due to a complex interaction between the host immune system and environmental triggers, with multiple genes conferring potential risk. A genetic component of sarcoidosis is recognized due to a markedly increased rate of disease in monozygotic twins compared to dizygotic twins, with a strong relative risk of 4.7 for disease in family members of affected patients. Multiple HLA alleles may play a role in conferring a predilection for developing the disease, and the genes involved may vary by ethnic group. HLA-DRB1 ∗ 01 and DRB1 ∗ 04 may be protective, while DRB1 ∗ 03, DRB1 ∗ 11, DRB1 ∗ 12, DRB1 ∗ 14, and DRB1 ∗ 15 may confer an increased risk. One subtype of sarcoidosis, Löfgren’s syndrome, characterized by hilar adenopathy, fever, arthritis, and erythema nodosum, which tends to be acute and self-resolving, may be associated with HLA-B8/DR3. African-American patients, an ethnic group with some of the highest overall incidence of sarcoidosis and a tendency toward chronic disease, may be more impacted by the HLA-DQB1 alleles. Beyond the HLA alleles, specific genes may confer disease susceptibility, including polymorphisms in the TNF gene and the IL23R gene; polymorphisms in the gene encoding angiotensin-converting enzyme have also been identified in patients with sarcoidosis. While sarcoidosis has historically been considered a Th1-predominant immune reaction, the granulomatous inflammatory cascade likely involves both the innate immune system (via Toll-like receptors) and potentially the Th17 immune system as well. The key cytokines include Th1 cytokines such as IL-2, IL-12, IL-18, and IFN-γ, granulocyte–monocyte colony stimulating factor, and TNF-α production by macrophages. The inflammatory pattern may vary by ethnicity, inciting antigenic trigger, or potentially disease phenotype, with different patterns of inflammation in patients with acute and self-resolving sarcoidosis compared to patients with chronic inflammatory or fibrotic disease. The end result is that sarcoidosis is a multifactorial combination of possible immunologic (T-cell receptor), genetic (HLA), infectious, or environmental factors (antigen[s]), and to date no single component can explain all cases of sarcoidosis.
Because of the lack of a consistent identifiable antigen(s), the polymorphic nature of the disease, and the lack of an animal model, a definitive pathogenesis is speculative at present and active research is ongoing. Sarcoidosis is believed to be a disorder of the lymphoreticular system characterized by the depression of cell-mediated immunity (delayed-type hypersensitivity), an imbalance of CD4/CD8 cells (helper-to-suppressor T-cell ratio), a hyperreactivity of B cells, and increased production of circulating immune complexes. It is believed that the interaction between T cells and antigens, antigen-presenting cells and cytokines in the correct genetic milieu affects T-cell function, which results in a secondary increase in B-cell activity. Prolonged antigenic stimulation may affect macrophages. As sarcoidal granulomas develop, enzyme secretion (in particular, secretion of angiotensin-converting enzyme—ACE) may occur. Elevation of ACE is probably the result of granuloma formation, and in some patients may reflect overall granuloma burden, although the ACE level overall lacks sensitivity, specificity, or reliability and cannot readily be utilized to either diagnose or follow disease activity over time. The precise reasons for the effects of the presumptive causative antigen(s) on patients with sarcoidosis are not understood. However, the result appears to be an overall somewhat anergic state, with a reduced reaction to recalled antigens, such as purified protein derivative, Candida , mumps, and the inability to sensitize an individual with sarcoidosis to an antigen such as dinitrochlorobenzene. Elevated levels of γ-globulin and nonspecific elevation of antibody titers to various viruses and fungal elements may also occur.
Clinical Manifestations
Cutaneous Manifestations
The skin is involved in 25% to 30% of patients with sarcoidosis, and the cutaneous manifestations of sarcoidosis are protean. Individual skin lesions are classified as either specific or nonspecific, based on the histopathologic examination. Those cutaneous lesions represented histopathologically by noncaseating granulomas are termed “specific” cutaneous lesions of sarcoidosis. Those lesions that do not show noncaseating granulomas are termed nonspecific, and are usually applied to lesions such as erythema nodosum. The specific cutaneous lesions include papules, plaques, nodules, subcutaneous nodules (“Darrier–Roussy” sarcoidosis), scar— or tattoo—associated papules, and lupus pernio. In addition, there are multiple less common presentations of specific cutaneous lesions such as acquired ichthyosis, erythroderma, psoriasiform, ulcerative, verrucous, scarring or nonscarring alopecia, photodistributed, and angiolupoid sarcoidosis. Interestingly, nail disease has been reported both as specific and nonspecific.
The most common nonspecific manifestation of sarcoidosis is erythema nodosum (EN). These lesions are firm, slightly erythematous to red-brown subcutaneous nodules, most commonly occurring symmetrically on the anterior tibial surfaces. They are often tender to palpation. When erythema nodosum is associated with sarcoidosis, it is frequently accompanied by uveitis, fever, arthritis, and asymptomatic bilateral hilar lymphadenopathy. This four-symptom complex is referred to as Löfgren’s syndrome. It is an acute form of sarcoidosis that self-resolves in 90% or more of affected individuals, although sometimes patients require transient anti-inflammatory treatment for symptom control. It is postulated that these patients have circulating immune complexes that cause erythema nodosum. Biopsies of the lungs and lymph nodes in these patients reveal sarcoidal granulomas. Biopsy is not indicated in patients who have the typical tetrad. Löfgren’s syndrome is most common in patients of northern European ancestry, and is relatively rare in African-Americans. Several other sarcoidosis syndromes have been described, including Heerfordt–Waldenström syndrome (fever, parotid gland enlargement, anterior uveitis, and facial nerve palsy); Mikulicz syndrome (bilateral sarcoidosis of the lacrimal, sublingual, submandibular, and parotid glands); and sarcoidosis–lymphoma syndrome (Hodgkin or non-Hodgkin lymphoma developing in patients with chronic active sarcoidosis).
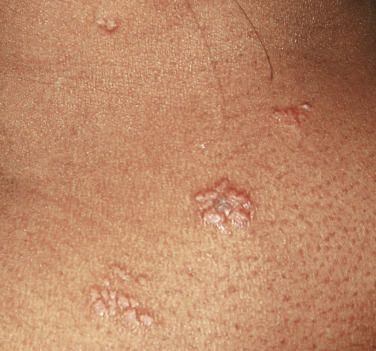
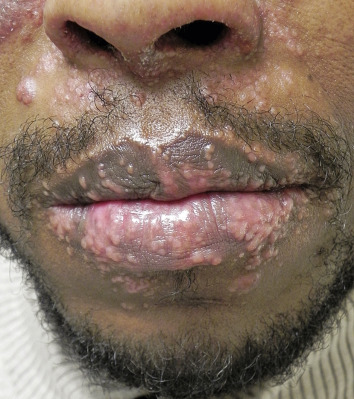
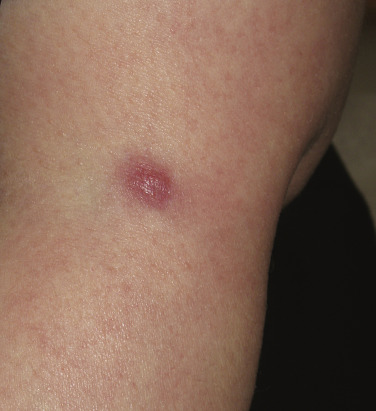
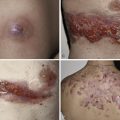
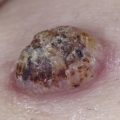
The “specific” cutaneous lesions of sarcoidosis occur in roughly 10% to 25% of patients with documented systemic sarcoidosis. The diagnosis of a cutaneous sarcoidal granuloma is made by skin biopsy. Diascopy ( Fig. 36-1 ) may help the dermatologist consider a granulomatous disease process, revealing “apple-jelly” orange-brown colors, but is not exclusive to sarcoidosis. Certain cutaneous lesions may be highly suggestive of sarcoidosis, but none are clinically diagnostic. The cutaneous lesions of sarcoidosis can mimic many dermatoses and should frequently be included in differential diagnoses, especially the other noninfectious granulomatous diseases granuloma annulare, necrobiosis lipoidica, necrobiotic xanthogranuloma, actinic elastolytic granuloma, reactive granulomatous eruptions such as interstitial granulomatous dermatitis and palisaded neutrophilic granulomatous dermatitis, cutaneous Crohn’s disease, cheilitis granulomatosis, foreign-body reactions, and the granulomatous facial dermatitidies, as well as infectious granulomatous diseases such as deep fungal infections, atypical mycobacteria, leprosy, syphilis, and cutaneous tuberculosis or cutaneous tuberculid reactions to remote tuberculosis infections. The most common cutaneous presentation of sarcoidosis is a papular lesion: small papules, erythematous-to-violaceous, 3 to 5 mm in diameter, are frequently noted on the head and neck ( Fig. 36-2 ). The periorbital and periorificial regions are commonly involved, with lesions commonly seen around the nostrils on the nares, and around the eyes and/or mouth ( Fig. 36-3 ). The lesions may be flesh-colored, red, violaceous, or slightly hyperpigmented. At times the papules enlarge or coalesce to form annular lesions ( Fig. 36-4 ), nodules ( Fig. 36-5 ), or plaques ( Fig. 36-6 ). Papular disease at the corners of the mouth may often coalesce and split, being indistinguishable from the classic split papule of secondary syphilis.
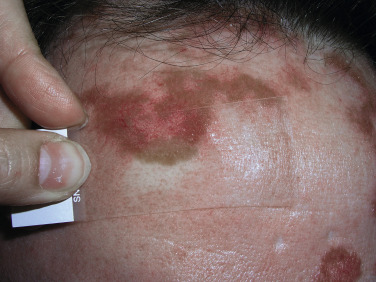
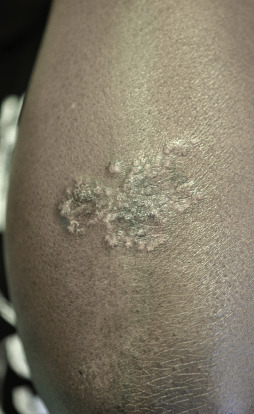
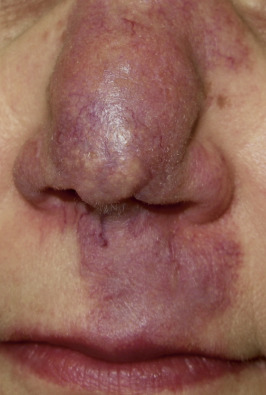
Indurated plaque-type lesions are also common, and can involve any area of the body. Infiltrated lesions involving the face, specifically the nose and cheeks, with or without scale, are classically referred to as lupus pernio ( Fig. 36-7 ). If extensive telangiectatic lesions are evident, this is termed angiolupoid sarcoidosis. Chronic longstanding lupus pernio and angiolupoid sarcoidosis can ulcerate, and patients with extensive nasal involvement have higher rates of sarcoidosis involving the upper respiratory tract (i.e., the sinuses), pulmonary fibrosis, and possibly bone cysts. Patients with lupus pernio or angiolupoid sarcoidosis frequently have chronic sarcoidosis, and the pulmonary involvement is often persistent. This cutaneous pattern is also often treatment refractory and requires aggressive therapeutic intervention to achieve partial or complete remission. Patients with papular, nodular, or plaque-like lesions on the face have been misrepresented innumerable times as having lupus pernio, and are not associated with a greater risk of pulmonary disease. In fact, all cutaneous disease manifestations other than EN have yet to be convincingly proven as having prognostic implications.
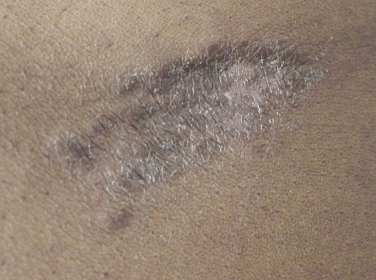
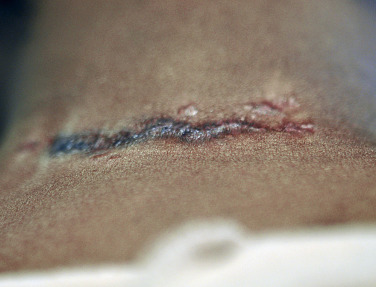
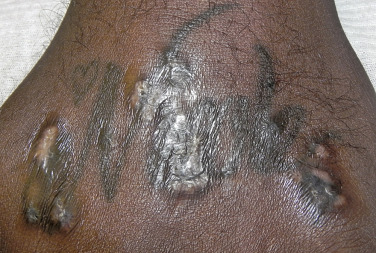
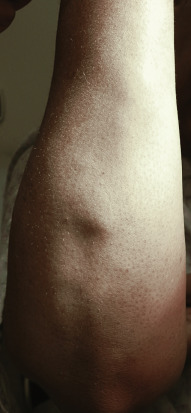
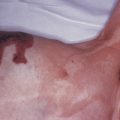
Sarcoidosis often occurs in scars ( Fig. 36-8 ), tattoos ( Fig. 36-9 ), or on areas of skin previously damaged by infection, radiation, or prior trauma. Sometimes it becomes difficult to determine whether these lesions are indeed cutaneous sarcoidosis, or whether they are local sarcoidal reactions to a foreign substance. Pathologic analysis of biopsies from patients with systemic sarcoidosis have demonstrated that foreign material is sometimes present (approximately 25% of the time) in the skin biopsy specimens. In the absence of systemic disease, a diagnosis of sarcoidosis cannot be made with confidence, particularly when only one area of the body is involved. The subcutaneous nodular lesions are often asymptomatic, and they frequently occur on the trunk and extremities ( Fig. 36-10 ). These lesions are rarely tender. A review of Mayo Clinic patients with subcutaneous sarcoidosis noted that such patients were more likely to be women in their fourth decade, and for there to be a strong association with bilateral hilar adenopathy, the relevance of which is uncertain given the high rates of pulmonary involvement (including hilar adenopathy) in sarcoidosis patients overall.
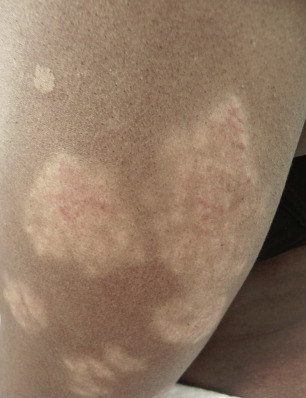
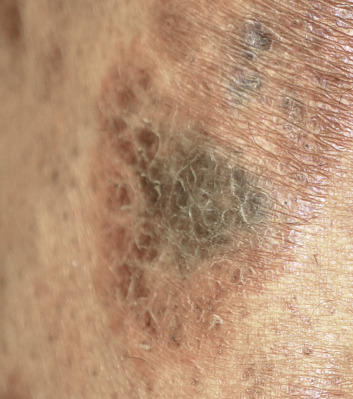
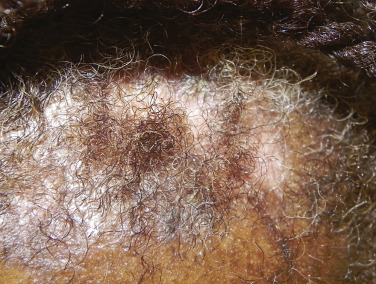
There are many unusual forms of sarcoidosis, as previously mentioned. Ulceration of sarcoid lesions is unusual, but can be seen on the lower extremities, where the disease may clinically and histologically mimic necrobiosis lipoidica. Verrucous lesions, although rare, occur most commonly in black females. They are reported most frequently on the lower extremities but may occur at any site. Macular hypopigmentation can rarely be seen and may be mistaken for hypopigmented mycosis fungoides or leprosy ( Fig. 36-11 ). Ichthyosis-like lesions occur in many patients with sarcoidosis ( Fig. 36-12 ); while there is a broad differential for causes of acquired ichthyosis, biopsies of the ichthyotic skin in patients with sarcoidosis may reveal typical granulomatous inflammation. Sarcoidosis may also affect the orogenital mucosa or the nail unit, and can cause a scarring or nonscarring alopecia ( Fig. 36-13 ). Erythrodemic presentations of sarcoidosis are rare but have been reported. Sarcoidosis has not been reported to present with either bullae or pustules.
Intrathoracic Disease
Intrathoracic disease, including hilar adenopathy and pulmonary parenchymal disease, is the most common manifestation of systemic sarcoidosis, occurring in more than 90% of patients. Many patients are asymptomatic; symptoms may include dyspnea, a chronic, dry, nonproductive cough, or severe breathing dysfunction. Pulmonary sarcoidosis can be staged, according to chest X-ray findings, from 0 to III. Stage 0 disease consists of no changes seen on chest X-ray studies. Stage I includes bilateral hilar adenopathy in the absence of parenchymal disease. Stage II consists of hilar adenopathy with the presence of interstitial pulmonary fibrosis. Stage III consists of extensive pulmonary fibrosis in the absence of hilar adenopathy. The stages of radiographic findings do not represent chronological development. A diagnosis of sarcoidosis is often made on a “routine” chest X-ray in an entirely asymptomatic patient. Patients who have bilateral hilar adenopathy without evidence of symptoms require monitoring; often the disease resolves spontaneously in all stages of pulmonary sarcoidosis. Abnormalities may be found by careful testing with pulmonary function tests and/or with bronchoalveolar lavage or ultrasound-guided transbronchial biopsies. Techniques such as positron emission tomography and high-resolution computed tomography (CT) scanning are also used.
Bilateral hilar adenopathy is the earliest and most common intrathoracic manifestation of sarcoidosis. Patients with this finding are often asymptomatic, although erythema nodosum, uveitis, arthritis, and fever may accompany the hilar lymphadenopathy, as previously mentioned. These patients with stage I disease rarely develop progressive parenchymal disease. Biopsy is not necessary in most patients with bilateral hilar adenopathy with symptoms of Löfgren’s syndrome, or in those who are asymptomatic. However, only a small proportion of patients who have either unilateral hilar adenopathy or dyspnea have sarcoidosis. The differential diagnosis of bilateral hilar adenopathy includes deep fungal infection, tuberculosis, lymphoma, and bronchogenic carcinoma. Patients with these manifestations should undergo a lung biopsy and, possibly, mediastinal nodal biopsies. Stages II and III pulmonary sarcoidosis are associated with a much higher incidence of chronic progressive disease, and resolution of the X-ray findings becomes less likely. Symptoms may be present or absent. However, abnormal pulmonary function testing correlates with more severe disease seen on chest X-ray studies, and forced vital capacity is typically followed for disease progression and in clinical trials to measure sarcoidosis response to treatment.
Ocular Manifestations
Ocular involvement occurs in approximately one-quarter of patients with sarcoidosis, and may affect any portion of the eye. Acute ocular sarcoidosis generally runs a course of 2 years or less, during which time active therapy may be needed. Chronic eye disease is less common. Early aggressive intervention can prevent scarring and blindness.
The most common ocular manifestation of sarcoidosis is uveitis, which usually affects the anterior segment of the eye. Patients may be entirely asymptomatic, or they may present with a red eye, photophobia, or increased tear production ( Fig. 36-14 ). The diagnosis is made by slit-lamp examination, which reveals “mutton fat” keratic precipitates in the anterior chamber. Less commonly, the posterior segment of the eye is involved, and its appearance correlates well with chorioretinitis and neurosarcoidosis. Uveitis must be aggressively treated to prevent adhesions, with resulting glaucoma, cataract development, or blindness. As ocular sarcoidosis may be clinically asymptomatic, up to the point of severe visual compromise, patients must undergo regular screening with at least annual ophthalmological exams.
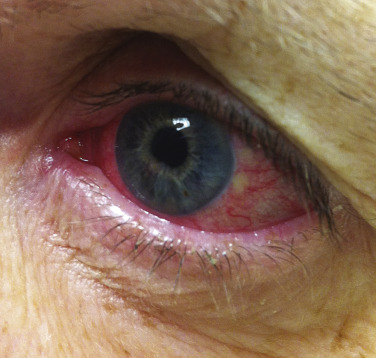
Another common ocular finding in patients with sarcoidosis is conjunctival and lacrimal gland involvement. This may or may not produce symptoms of ocular irritation. Conjunctival granulomas occur in about one-third of patients with sarcoidosis. Conjunctival biopsy specimens may be positive even when the patient lacks clinical evidence of conjunctival involvement.
Lymph Nodes
Sarcoidal granulomas commonly infiltrate the lymph nodes. The incidence of peripheral lymphadenopathy is roughly 30% in patients with systemic sarcoidosis. Lymphadenopathy is associated with both acute and chronic disease patterns. Lymph node involvement, detected by palpation, is usually nontender, and is often not noticed by the patient. Because of the increased risk of lymphoma in patients with sarcoidosis (termed the sarcoidosis–lymphoma syndrome), extensive adenopathy or new adenopathy presenting out-of-step with a patient’s sarcoidosis should be evaluated with fine-needle aspiration or biopsy of lymph nodes, as histologically they can be easily distinguished.
Splenic involvement in sarcoidosis is detected in approximately 20% to 25% of patients. It is manifested by splenomegaly, but functional abnormalities are rare.
Musculoskeletal Manifestations
Symptomatic muscle involvement in sarcoidosis is rare, although biopsy-based detection of a sarcoidal infiltrate is not uncommon. Clinical series of large numbers of patients with sarcoidosis reveal that approximately 1% have symptoms of muscle involvement. However, when patients with sarcoidosis undergo random muscle biopsies, more than 50% have histologic evidence of granulomatous disease. At times, muscle involvement may appear as subcutaneous nodules, although this is rare.
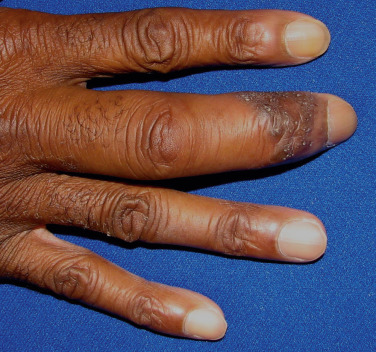
Bone lesions occur in 10% to 15% of patients, usually correlating with chronic progressive disease. The X-ray changes are cystic lesions that usually occur in the terminal phalanges of the hands. These may be accompanied by soft tissue swelling ( Fig. 36-15 ). Arthralgias are commonly experienced in acute sarcoidosis, particularly in patients with Löfgren’s syndrome, and may be accompanied by arthritis. The wrists, knees, and ankles are the most commonly affected joints. Although chronic granulomatous arthritis has been reported, it is a rare complication of systemic sarcoidosis.
Neurosarcoidosis
Neurosarcoidosis affects 5% to 15% of patients with systemic sarcoidosis. Central nervous system involvement occurs in 50% of patients with neurosarcoidosis. The presence of neurosarcoidosis correlates with the presence of posterior uveitis and chronic cutaneous disease. The most common neurologic manifestations of sarcoidosis include cranial neuropathies such as optic nerve disease or facial nerve palsy, meningitis, and cerebral granulomas, which can lead to encephalopathy and seizures. Heerfordt–Waldenström syndrome is also frequently associated with central nervous system involvement. The detection of neurosarcoidosis can be difficult because affected tissue is not readily available for microscopic evaluation. The evaluation of a patient with suspected neurosarcoidosis can include skull X-ray studies, electroencephalography, a CT scan, and/or magnetic resonance imaging of the brain. Lumbar puncture may also be useful in patients with meningeal involvement. Small-fiber neuropathy can occur due to sarcoidosis and can present with peripheral nerve symptoms, including atypical pain.
Hepatic Sarcoidosis
Hepatic involvement in sarcoidosis is common. In a large study, hepatomegaly was detected in 20% to 50% of patients with sarcoidosis. Abnormal liver function tests are present in approximately half of patients with hepatic involvement. Liver biopsy may be useful to obtain tissue to establish a diagnosis of sarcoidosis, although care must be taken to exclude other causes of granulomatous disease of the liver, and the findings must be correlated with evidence of sarcoidal disease elsewhere in the body. Hepatic involvement rarely progresses to functional abnormalities or cirrhosis. Combined hepatosplenomegaly is associated with a poorer outcome owing to the higher overall body granuloma and fibrosis burden.
Endocrine, Metabolic, and Laboratory Abnormalities
Endocrine glands may be infiltrated by sarcoidal granulomas. Functional abnormalities are not common, although pituitary or hypothalamic infiltration can cause diabetes insipidus or, on rare occasions, panhypopituitarism. An elevated prolactin level is a useful indicator of hypothalamic sarcoidal involvement. A number of studies have indicated a potential increased risk of thyroid disease in patients with sarcoidosis, although it is not clear if that is due to direct granulomatous inflammation or shared autoimmune inflammation. Other endocrine organs, such as the parathyroid and adrenal glands and the pancreas, may be involved. However, functional impairment of these organs is unusual.
Hypercalcemia occurs in some patients with sarcoidosis. In occasional patients with widespread sarcoidosis, the serum calcium level elevation can be persistent, and can lead to urinary tract stones, nephrocalcinosis, and even renal failure. Hypercalciuria is more common than hypercalcemia and may be used as a correlate with disease activity. The granulomas in sarcoidosis are metabolically active and produce high levels of 1α-hydroxylase, converting 25-hydroxy vitamin D to its active 1,25-dihydroxy form, and leading to increased vitamin D activity and elevated serum calcium.
The serum ACE level is raised in approximately 60% of patients with systemic sarcoidosis. ACE is produced by epithelioid cells and may reflect the granuloma load in the body. ACE levels are neither sensitive nor specific to sarcoidosis, and medications and other pulmonary inflammatory processes may lead to elevations. When the ACE level is markedly elevated, two to three times the upper limit of normal, sarcoidosis may be more likely. ACE is not routinely reliable in monitoring disease activity, but may trend with granuloma burden in a subset of patients. Other blood levels, such as serum IL2 receptor or chitotriosidase, are investigational and are not widely available or clinically practical in evaluating patients with systemic sarcoidosis.
Cardiac Disease
The true incidence of cardiac involvement in sarcoidosis is not known. However, autopsy studies suggest that it is more common than clinically apparent and frequently asymptomatic. Cardiac sarcoidosis can result in symptoms of congestive heart failure, arrhythmia, or conduction defects. The most common symptom is sudden death. Appropriate screening for cardiac sarcoidosis is controversial and recommendations are rapidly changing. All patients should have a thorough review of symptoms and electrocardiography; any abnormalities (including a history of palpitations) likely warrant further testing, such as 24-hour Holter monitoring, echocardiogram, cardiac positron emission tomography, magnetic resonance imaging, or referral to a cardiologist.
Other Clinical Manifestations of Sarcoidosis
Almost any area of the body can be affected by sarcoidosis. Granulomatous renal disease has been reported on several occasions, although renal impairment is more often due to persistent hypercalcemia and hypercalcuria. Gastric granulomas, breast soft tissue granulomas, bone marrow granulomas, spinal cord lesions, and gonadal granulomas have also been reported.
Relationship of Cutaneous Disease to Systemic Disease
Many studies detail the cutaneous disease in patients with sarcoidosis. Unfortunately, none of these studies used the same methods, and many did not define the cutaneous lesions adequately. However, a few generalizations can be made. Patients with lupus pernio more frequently have sarcoidosis of the upper respiratory tract and lungs, as well as possibly more bone involvement, than do patients without lupus pernio. Those patients with EN and bilateral hilar lymphadenopathy (Löfgren’s syndrome) have a self-limiting course and a good prognosis. In all cases of cutaneous sarcoidosis, patients require evaluation for systemic involvement.
Histopathologic Findings
Sarcoidosis is characterized by granulomas composed principally of epithelioid cells with an occasional giant cell and little or no caseation necrosis, with scant perigranulomatous lymphocytic inflammation. Inclusion bodies (Schaumann, Hamazalki–Wasserman, and asteroid bodies) may be observed in varying numbers, but these are not specific for sarcoidosis and may occur in other granulomatous conditions. The granulomas may remain seemingly unchanged for months or years, may resolve completely, or may undergo a fibrotic change. Although this is classically a dermal disease, epidermal change has been demonstrated histologically (particularly in vulvar sarcoidosis, wherein transepidermal elimination of granulomas has been reported).
The histopathologic differential diagnosis includes other granulomatous diseases, which may be excluded by special stains (infectious granulomas) and/or examination for foreign material (foreign body granulomas). Because noncaseating granulomas are not specific for sarcoidosis, other conditions must be vigorously excluded before a diagnosis of sarcoidosis can be confirmed. Thus, the diagnosis is one of exclusion. Special stains of the histopathologic specimens should include the acid-fast bacilli and Fite stain for mycobacteria, and periodic acid–Schiff stain for fungal elements. In addition, examination for a foreign body reaction with at least polarized light should be undertaken, although the presence of foreign material does not exclude the diagnosis, with refractile material reported in up to 25% of sarcoidosis skin biopsies. Various neoplasms should be excluded, particularly lymphomas, because there are occasional reports of sarcoidal tissue reactions occurring in nodes adjacent to neoplastic change.
Diagnosis and Differential Diagnosis
Sarcoidosis is diagnosed by a combination of clinical, radiologic, and laboratory findings plus the demonstration of noncaseating granulomas in the tissue. The differential diagnosis varies according to the organs involved. The differential mainly revolves around infectious and noninfectious granulomatous disease processes for each organ system, and has been discussed above. The only exception is the diagnosis in patients with asymptomatic bilateral hilar lymphadenopathy or in patients who have Löfgren’s syndrome. These two clinical presentations are characteristic enough not to require histopathologic confirmation, but require appropriate follow-up to ensure resolution of the disease process. Tissue diagnosis through biopsies of various sites remains the main procedure to confirm the diagnosis of sarcoidosis. A prime tissue for biopsy is the skin, because it is an accessible, high-yield organ. Particular attention should be paid to changes in scars, tattoos, or to any papular, nodular, or plaque-type lesion of recent onset, particularly on the nose, around the nostrils, or around the eyes and mouth. In addition, conjunctival biopsy, even in the absence of conjunctival nodules, may be positive in up to one-third of patients with sarcoidosis. Palpable lymph nodes may be biopsied when feasible. Lung biopsy through a fiberoptic bronchoscope is also helpful in establishing a diagnosis. Mediastinoscopy may be used to approach intrathoracic nodes not accessible via bronchoscopy. Blind biopsy of the minor salivary glands on the lower lip is said to be positive in up to 60% of patients with sarcoidosis. Liver biopsy is a high-yield procedure, but its morbidity and mortality rates make it less useful than the previously mentioned techniques. Muscle biopsy may reveal sarcoidal granulomas in up to 50% of patients, particularly when a needle is used. Other less accessible sites for biopsy include the bone marrow, kidney, and spleen when involvement is suspected.
Evaluation
After a diagnosis of sarcoidosis is made, the patient should be thoroughly evaluated to define the organ systems involved and to aid in prognostic predictions and therapeutic decisions. A team approach with the internist, ophthalmologist, pulmonologist, and dermatologist is required. A careful history and physical examination should be performed, focusing on all organ systems, but with preference given to the lungs, eyes, nervous system, heart, and skin. Laboratory blood testing for chemistries, renal function, liver enzymes, and calcium level should be done. An electrocardiogram is required, and further cardiac evaluation is indicated if there are elements of the patient’s history, symptoms, or electrocardiogram abnormalities. Chest imaging studies and pulmonary function tests with diffusion studies should be ordered, and should be repeated on an annual basis. All patients should undergo ophthalmological evaluation at the time of diagnosis and annually. In patients with persistent hypercalcemia or a history of kidney stones, careful evaluation of the urine is important, which may include 24-hour urine calcium levels. Additional testing could consist of skin tests to detect anergy, purified protein derivative, or interferon-γ release assay tests, creatine kinase/aldolase for suspected muscle disease, urinalysis, quantitative immunoglobulins, and protein electrophoresis. The measurement of serum ACE levels is occasionally helpful in following patients with pulmonary sarcoidosis, but not routinely for skin disease. Vitamin D levels should be evaluated, with testing for both the 25-hydroxy and 1,25-dihydroxy forms indicated. Given the potential association of sarcoidosis with thyroid disease, screening for thyroid function testing is reasonable.
Prognosis
Mortality rates in sarcoidosis vary from 3% to 6%. Cardiac disease (the most common cause in Japan), progressive pulmonary disease (the most common cause in the United States), and neurosarcoidosis have been the cause of death in some patients.
The morbidity in sarcoidosis can be severe. Blindness can result from untreated ocular disease, and pulmonary disease can cause debilitating fatigue and shortness of breath, sometimes requiring oxygen supplementation or, in severe cases, lung transplantation. Renal failure requiring dialysis has been reported from granulomatous involvement of the kidney, calcium deposits, and chronic urinary tract stones. Cosmetic deformities may occur in patients with cutaneous sarcoidosis, particularly in those with lupus pernio. Although many patients with pulmonary sarcoidosis may have resolution of their disease, the rates vary with the stage of pulmonary disease. Stage I sarcoidosis resolves in roughly 60% of patients; stage II resolves in 40% of patients; and stage III resolves in only 12%.
Despite the aberrations noted in tests of cell-mediated immunity, untreated sarcoidosis is not associated with an increased number of infections. However, it has been linked to an increase in the frequency of malignancy, although some recent meta-analyses have cast doubt on these associations and suggest there may not be an increased risk of malignancy in patients with sarcoidosis. Prior studies had suggested an increased risk of skin cancers (nonmelanoma and melanoma), and several studies have reported an increased incidence of hematologic malignancy—i.e., lymphoma, leukemia, and solid tumors (i.e., lung, testes, skin). Of note is that smoking has been associated with a reduced risk of developing pulmonary sarcoidosis because of its ability to impair the pulmonary immune function, but for obvious reasons patients should not be encouraged to smoke. It is not clear, however, from these studies whether sarcoidosis is the primary disease, or whether a local sarcoidal reaction is a response to the malignancy in this group of patients. Of interest is the induction of new-onset sarcoidosis with antineoplastic agents used to treat oncology patients with Hodgkin and non-Hodgkin lymphoma, and the biologic modifier α-interferon (IFN) in leukemia. This has also been reported in noncancer patients who have received IFN-α for chronic hepatitis C. Drug-induced sarcoidosis has also been reported in patients receiving TNF-α inhibitors for diseases such as psoriasis.
Treatment
Acute sarcoidosis in which bilateral hilar adenopathy exists alone or in combination with erythema nodosum, uveitis, or arthritis is usually a self-limited disease and does not require specific therapy. Symptomatic therapy for the erythema nodosum or arthritis could include nonsteroidal anti-inflammatory drugs, such as aspirin or indometacin. Acute uveitis can be treated with corticosteroid eye drops. Some patients with severe symptoms require brief courses of oral corticosteroids.
There are no Food and Drug Administration-approved treatments for cutaneous sarcoidosis, and the level of evidence supporting various off-label options is generally limited to case series and retrospective analysis. However, there exists expert opinion and consensus regarding an overall approach, starting with topical corticosteroids and progressing to immunomodulatory and immunosuppressive options. Chronic cutaneous lesions, particularly lupus pernio and indurated plaques, which can cause scarring and disfigurement, should be treated more aggressively. Topical corticosteroids and topical immunomodulators are variably effective. Topical steroids should be selected on a site-specific basis to avoid risks of atrophy and dyspigmentation. Intralesional corticosteroids are generally more effective, and in some lesions may lead to a durable response, at rates ranging from 5 to 40 mg/cc of intralesional kenalog.
Antimalarials, particularly hydroxychloroquine sulfate (200 to 400 mg/day) or chloroquine phosphate (250 mg/day), are useful in treating patients with cutaneous sarcoidosis, and should be considered first-line therapy for patients with cutaneous sarcoidosis that is not responsive or too widespread to be amenable to topical steroids. Antimalarial therapy requires ophthalmologic monitoring; however, patients with sarcoidosis should be followed routinely by ophthalmologists regardless. Antimalarials have no effect on most extracutaneous manifestations of sarcoidosis. Tetracycline class antibiotics, particularly minocycline (100 mg twice daily), may also be effective in treating cutaneous sarcoidosis.
Methotrexate (10 to 15 mg/week) is the next-line therapy to supplement or replace the use of antimalarials. Methotrexate is highly effective in treating cutaneous sarcoidosis and is furthermore frequently used to treat extracutaneous disease. Corticosteroids are another option for patients with severe cutaneous disease, but most patients will demonstrate transient disease improvement while on steroids and then relapse as the drug is tapered. Thalidomide is an alternative option to methotrexate in patients who require treatment beyond antimalarials; patients must be enrolled in a monitoring program, and the rates of side effects, particularly neuropathy, are significant.
In patients with chronic cutaneous sarcoidosis such as lupus pernio, the TNF-α inhibitors have shown promise, with excellent tolerability and response rates in patients with previously refractory disease. Adalimumab and infliximab have the most data, and etanercept was not effective in a small trial.
Other options include pentoxifylline, apremilast, azathioprine, cyclophosphamide, cyclosporine, chlorambucil, leflunomide, isotretinoin, melatonin, fumaric acid esters, and allopurinol, which have been described as beneficial in anecdotal reports or small case series for treating cutaneous sarcoidosis. Nonmedical therapies have included surgical procedures such as the use of lasers, dermabrasion, surgical excision with grafting, plastic surgery, and phototherapy (narrowband ultraviolet B phototherapy, ultraviolet A-1 phototherapy, and photodynamic therapy), but there is not enough evidence to recommend any of these as the standard of care, and destructive modalities should be used with caution as sarcoidosis may develop at sites of trauma.
Ocular sarcoidosis, which can lead to scarring and blindness, must be aggressively treated. Corticosteroid eye drops may be effective; however, patients who do not respond or who respond only partially may require intraocular corticosteroid injections or systemic corticosteroid therapy.
Progressive pulmonary disease is considered to be an indication for systemic corticosteroid therapy. Documented changes in pulmonary function tests as a result of therapy have been reported. Alternative therapies that may be effective or that reduce the corticosteroid dosage include various immunosuppressive agents, particularly methotrexate. However, too few studies are available to reliably evaluate the effects of any agent other than systemic corticosteroids.
In addition to chronic disfiguring cutaneous lesions, ocular lesions, and progressive pulmonary disease, the indications for systemic corticosteroid therapy include hypercalcemia, neurosarcoidosis, symptomatic cardiac sarcoidosis, and functional endocrinologic abnormalities.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


