Abstract
Oncogenes act in a dominant fashion and gain-of-function results in increased cellular proliferation. Tumor suppressor genes act in a recessive fashion and loss of normal function results in uncontrolled growth. TP53 , the tumor suppressor gene that encodes p53, is the single most frequently mutated gene in human cancer and controls signaling pathways involved in cell division and apoptosis. Mutations in either apoptosis- or senescence-regulating genes are considered “driver” mutations and are uniformly present in cancers.
Disruption of the normal function of the Sonic hedgehog (SHH)–Patched–Smoothened (SMO) pathway is required for the development of basal cell carcinoma (BCC). BCCs, the most frequent cancer in humans, develop within skin that contains hair follicles, and they arise without precursors. BCCs are stroma-dependent, locally invasive, and very rarely develop metastasis.
Squamous cell carcinomas (SCCs) arise in chronically sun-exposed skin and originate from epidermal keratinocytes. SCC develops through a series of progressive stages, including actinic keratosis, SCC in situ , invasive cancer, and eventual metastasis.
Keywords
basal cell carcinoma, squamous cell carcinoma, p53, TP53 , Patched, Sonic hedgehog (SHH), Smoothened, Hedgehog signaling pathway, oncogenes, tumor suppressor genes, apoptosis, skin cancer, PTCH1
- ▪
Oncogenes act in a dominant fashion and gain-of-function results in increased cellular proliferation
- ▪
Tumor suppressor genes act in a recessive fashion and loss of normal function results in uncontrolled growth
- ▪
TP53 , the gene that encodes p53, is the single most frequently mutated gene in human cancer; p53 controls signaling pathways involved in cell division and apoptosis
- ▪
Mutations in either apoptosis- or senescence-regulating genes are considered “driver” mutations and are uniformly present in cancers
- ▪
PTCH1 controls proliferation and differentiation, and disruption of its normal function is required for the development of basal cell carcinoma (BCC)
- ▪
BCCs, the most frequent cancer in humans, develop in skin that contains hair follicles, and they arise without precursors
- ▪
BCCs are stroma-dependent, locally invasive, and very rarely develop metastasis
- ▪
Squamous cell carcinomas (SCCs) arise in chronically sun-exposed skin and originate from epidermal keratinocytes
- ▪
SCC develops through a series of progressive stages, including actinic keratosis, SCC in situ , invasive cancer, and eventual metastasis
Introduction
No definition of cancer is entirely satisfactory from a cell biology point of view, despite the fact that cancer is essentially a cellular disease, characterized by a transformed cell population with autonomous cell growth and invasive behavior. Microscopic evaluation of a tissue section taken from an excised skin tumor remains the gold standard for determining the diagnosis of skin cancer. Analysis of genomic DNA, transcribed genes, and expressed proteins adds important information to the histologic features detected by light microscopy. In the future, diagnosis and prognostic information as well as choice of treatment will most likely be based upon a synoptic evaluation of tissue architecture in conjunction with an analysis of molecular characteristics. Even today, evolving knowledge based on the human genome sequence and biochemical pathways, including signaling within and between cells, enables us to dissect some of the mechanisms that underlie different stages in tumor formation as well as the variation of phenotypes which define the different types of cancer.
Carcinogenesis
Malignant transformation represents the transition from a normal to malignant phenotype and is based on the progressive accumulation of genetic alterations. Although not formally proven, malignant transformation takes place in one cell from which a subsequently developed tumor originates (the dogma of cancer clonality). Carcinogenesis is the process by which cancer is generated and is generally accepted to include multiple events, which ultimately lead to growth of a malignant tumor . This multistep process includes several rate-limiting steps, representing the acquisition of mutations and epigenetic modifications, eventually leading to the formation of cancer (after various stages of precancerous proliferation).
The most common forms of cancer arise in somatic cells and are predominantly of epithelial origin (most often skin, prostate, breast, lung or colon), followed by cancers originating from hematopoietic lineage (leukemia and lymphoma) and mesenchymal cells (sarcomas). The stepwise changes involve an accumulation of errors (mutations) in vital regulatory pathways that control cell division, apoptosis, senescence, cell–cell and cell–matrix interactions, and cell death. Each of these changes provides a selective growth advantage compared to surrounding cells, resulting in a net growth of the tumor cell population. A certain degree of genomic instability is likely required in order to accumulate a sufficient number of mutations. The spontaneous mutation rate is not high enough to explain the speed with which a cancer develops and thus tumor cells display a “mutator phenotype” with a higher mutation rate compared to neighboring normal cells. Decreased efficiency in DNA repair systems is one important mechanism that leads to a “mutator phenotype” .
Knowledge of the events involved in carcinogenesis has been obtained from experimental cell culture and animal studies, as well as from clinicopathologic studies in humans. Information regarding cellular, molecular and genetic changes that represent tumor initiation and progression can be correlated with histopathologic findings. Primary genetic alterations leading to initiation of cancer can be identified and validated in experimental systems, but in clinical investigations this is not feasible. Progression is ascribed to the events that occur after malignant transformation and can in part be defined by histology. This multistep model includes defined stages of tumor development accompanied by features including multiple genetic and epigenetic events involving different signaling pathways. Specific changes in DNA, signifying different steps in cancer development, were first described for colorectal cancer . This model illustrates distinct genetic hits acquired during the passage from normal epithelium to metastatic cancer and provides support for the multi-hit carcinogenesis hypothesis.
The hallmarks of cancer have also been described from a functional perspective and consist of a number of molecular, biochemical, and cellular traits that are shared by most human cancers. Six traits have been proposed and include self-sufficiency with regard to growth signals, insensitivity to anti-growth signals, evasion of apoptosis, limitless replicative potential, sustained angiogenesis, and tissue invasion/metastasis . Self-sufficiency with regard to growth signals can be achieved by activation of oncogenes. Loss-of-function of tumor suppressor genes results in uncontrolled proliferation due to insensitivity to anti-growth signals. Evasion of apoptosis can be accomplished via inactivation of the TP53 tumor suppressor gene and production of survival factors. Constitutively active telomerase can render limitless replicative potential. The production of vascular endothelial growth factors results in sustained angiogenesis, and inactivation of cellular adhesion molecules, e.g. E-cadherin, facilitates the cell migration necessary for tissue invasion and metastasis. The order in which these capabilities are acquired is not static and varies amongst different forms of cancer.
Several traits can be achieved through a single genetic change, whereas a certain trait may require several genetic alterations. For example, inactivation of the TP53 tumor suppressor gene can result in insensitivity to anti-growth signals, resistance to apoptosis, and increased angiogenesis. In addition, impairment of cell cycle control and apoptotic pathways contributes to the “mutator phenotype”. In individuals predisposed to cancer at an early age, certain traits result from alterations in the germline and are thus carried in every cell. Xeroderma pigmentosum (“mutator phenotype”; mutations in nuclear excision repair genes; Ch. 86 ), familial melanoma (defect in cell cycle control; CDKN2A [INK4A] mutations), Li–Fraumeni syndrome (defect in cell cycle control/apoptosis; TP53 mutations), and basal cell nevus syndrome (BCNS; defect in control of proliferation/differentiation; PTCH1 mutations) are examples of how mutations within single genes in the germline predispose one to the development of cancer.
The Cell Cycle
Normal cell growth and cell mass are influenced by several internal factors, including signals which regulate proliferation, differentiation, and cell death. These factors are in part triggered by blood supply and the external environment, including soluble molecules as well as matrix–cell and cell–cell contacts. Proliferation involves DNA replication and mitosis via a series of events termed the cell cycle ( Fig. 107.1 ). Three primary checkpoints (G 1 , G 2 and M), which act to ensure successful cell division, have been identified in the cell cycle . Regulation of these G 1 , G 2 and M transitions involves three major protein families: cyclins, cyclin-dependent kinases (CDKs), and cyclin-dependent kinase inhibitors (CKIs). CDKs regulate the phosphorylation of key proteins involved in cell cycle progression, e.g. the retinoblastoma (Rb) protein. The concentration and balance of cyclins versus CKIs in turn regulate CDK activity. CDKs are activated by mitogenic growth factors and are removed by ubiquitin-mediated proteolysis in a cyclic manner correlating with the different phases of the cell cycle.
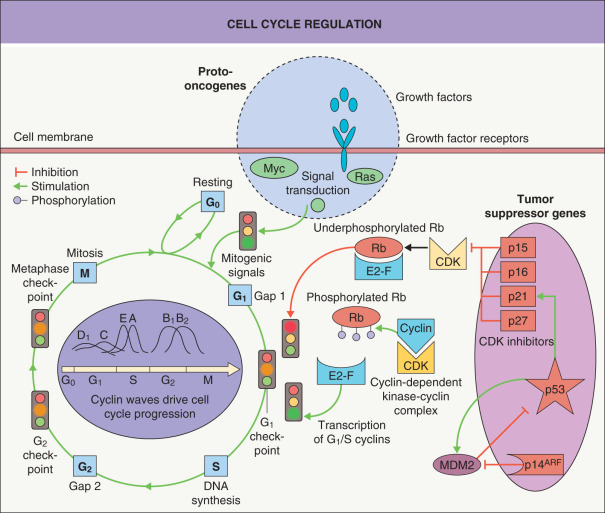
The G 1 transition is a critical one and is regulated by a complex interplay of macromolecules influenced by growth factors, hormones, and cell contacts. The key factor is the degree of phosphorylation of Rb. If, at the G 1 checkpoint, Rb is underphosphorylated, then cell proliferation is blocked and the cell is arrested in G 1 . The repression can be reversed by CDK–cyclin complex-mediated phosphorylation. This phosphorylation/dephosphorylation cycle can thus reversibly regulate cell cycle progression and rate of proliferation (see Fig. 107.1 ).
There are two major families of CKIs, and both are involved in the G 1 checkpoint. The family of inhibitors of CDK4, also referred to as INK4, consists of p15, p16, p18 and p19; these inhibitors specifically bind to CDK4 and CDK6. The other family of CKIs is less specific with respect to the type of CDK they bind and includes the general CDK–cyclin complex inhibitors p21, p27 and p57. Expression of these CKIs is in part tissue-specific.
The CDKN2A (INK4A) gene locus, which is involved in familial melanoma (see Ch. 113 ), has an extraordinary feature; it encodes two different mRNAs by shifting the codon reading frame (hence the term a lternative r eading f rame [ARF]) and these two mRNAs are independently regulated. As a result, the two protein products – p16 and p14 ARF – have different amino acid sequences and different functions. p16 is a CKI that blocks Rb phosphorylation, whereas p14 ARF binds MDM2, resulting in an increase in p53 through interference with the p53–MDM2 feedback loop (see Fig. 107.1 ). Thus, both p16 activation and p14 ARF activation lead to cell cycle arrest through different pathways and their dysfunction can lead to cell proliferation. In carcinomas, cyclin and CKI alterations are common, whereas activating mutations in CDKs are rare, e.g. only a few families with familial melanoma have been described with mutations in CDK4 . p53 and Rb alterations are also common events in human cancer.
Cancer Genes – Oncogenes and Tumor Suppressor Genes
With advances in technology, it is now possible to catalogue genome-wide changes in DNA, at unparalleled resolution. In breast and colon cancer, sequencing the exomes (coding exons) of tumors has revealed an average of 50 to 100 somatic mutations in “cancer genes” in each tumor . Are these all significant? The answer is probably no. Somatic alterations in tumor DNA are called either driver mutations that confer a selective growth advantage to the cancer cells or passenger mutations that have no effect on tumorigenesis. Somatic copy number alterations can also occur .
An increase in gene function (oncogenes) as well as a loss of gene function (tumor suppressor genes) will affect cell proliferation and can lead to uncontrolled growth. These two categories of genes, and their encoded proteins, represent the functional features that drive carcinogenesis at a molecular level. Oncogenes, originally named with three-letter acronyms (e.g. ras, myc, src, fos ), are genes that gain oncogenic or transforming potential as a result of genetic changes . More than 400 “cancer genes” have been identified that participate in diverse regulatory pathways that control cell fate; examples are listed in Table 107.1 .
| EXAMPLES OF ONCOGENES AND TUMOR SUPPRESSOR GENES ASSOCIATED WITH CARCINOGENESIS | ||||
|---|---|---|---|---|
| Gene | Protein product | |||
| Name | Function | Chromosome | Location | Function |
| PDGFB | Oncogene | 22 | Extracellular | Platelet-derived growth factor (PDGF) |
| HRAS | Oncogene | 11 | Membrane | GTPase |
| SRC | Oncogene | 20 | Cytoplasm/membrane | Tyrosine kinase |
| RAF1 | Oncogene | 3 | Cytoplasm/membrane | Serine/threonine kinase |
| MYC | Oncogene | 8 | Nucleus | Transcription factor |
| FOS | Oncogene | 14 | Nucleus | Transcription factor |
| RB1 | Suppressor gene | 13 | Nucleus | Cell cycle regulator |
| TP53 | Suppressor gene | 17 | Nucleus | DNA repair, apoptosis |
| BCL2 | Suppressor gene | 18 | Mitochondria | Apoptosis |
Extracellular signals such as growth factors and extracellular matrix components determine if cells move from a quiescent state into a proliferative state. These signals are transmitted into the cell through transmembrane structures that have distinct features (e.g. receptors) and the signals are then propagated into the nucleus by a multitude of interacting pathways (i.e. signal transduction). Increased production of growth factors and growth factor receptors is frequently observed in cancer cells; this results in autocrine and paracrine loops that lead to enhanced cell proliferation . Upon ligand binding, growth receptors mediate signals via tyrosine kinases or serine/threonine kinases on the cytosolic side of the membrane (see Ch. 55 ). Cancer cells can also alter receptor activity via “activating” mutations in the genes that encode the receptors, thus generating constitutively active, ligand-independent receptor molecules (e.g. KIT in acral melanoma).
The ras proto-oncogene, which is mutated in at least 30% of human cancers, plays a key role in signal transduction pathways between the cell membrane and the nucleus, communicating signals to a number of effector pathways. The ras family members are H-ras, K-ras and N-ras , and the 21 kDa Ras protein is a GTP-binding protein with latent GTPase activity, active when bound to GTP and inactive when GTP is hydrolyzed to GDP. The ras oncogene acts as a multifunctional modulator capable of redirecting input signals from growth receptors to alternative pathways. The end result of the signal cascade following Ras activation is increased transcription due to an alteration in the quantity or function of nuclear transcription factors (see Ch. 113 ).
For oncogenes, a change in one of the two inherited alleles leads to a gain-of-function that is dominant (over that of the unaffected allele). In contrast, tumor suppressor genes act in a recessive manner, i.e. a mutation in one allele has no effect ( Fig. 107.2 ). The biologic consequence of a recessive mutation becomes apparent when the second, normal (wild-type) allele is lost. Loss of this allele is termed loss of heterozygosity (LOH) and represents alteration from a heterozygous to a homozygous state for a gene ( Fig. 107.3 ).


The tumor suppressor genes are of critical importance in human carcinogenesis. In contrast to the diverse interactions of oncogenes converging to stimulate growth, tumor suppressor genes act by inhibiting proteins that control cell cycle progression. Inactivation of both alleles of a suppressor gene (one of which may be inherited in an inactivated state as in familial cancer syndromes) is required to inactivate the normal repressive function. In normal cells, inactivation of the protein products of tumor suppressor genes is achieved via binding to other proteins or by phosphorylation. In tumor cells, inactivation is often due to mutations, insertions, and/or deletions (allelic loss; see Fig. 107.3 ).
The Rb protein serves as a classic example for examining the functions of tumor suppressor genes. Underphosphorylated Rb protein blocks proliferation in normal cells by binding to, and thereby inactivating, a transcription factor (E2F); E2F is necessary for propagation of the cell cycle (see Fig. 107.1 ). Serine/threonine phosphorylation of the Rb protein disrupts this binding, releasing E2F and enabling cell cycle progression. Genes that control the levels of Rb phosphorylation (e.g. CDKN2A ) act as tumor suppressor genes and are often inactivated in cancer by mutation or promoter methylation.
The TP53 gene is frequently inactivated by a point mutation, and as opposed to classic tumor suppressor genes (that act in a recessive manner), TP53 mutations can act in a dominant-negative manner (see Ch. 54 & Fig. 107.3 ). This is because the p53 protein is a tetrameric protein and oligomerization of a mutant allele product and a normal allele product results in an inactive protein.
MicroRNA (miRNA) is non-coding RNA that regulates translation and degradation of specific mRNAs (see Fig. 3.15 ). Dysregulation of miRNA expression has been associated with oncogenic transformation, and miRNAs whose actions mimic oncogenes or tumor suppressors have been identified in human cancer. For example, miRNA-31 has been identified as a metastasis suppressor .
Epigenetic change is a modification to DNA or associated histone proteins, without variation in DNA sequence. There are two major mechanisms of epigenetic modification, DNA methylation (addition of a methyl [CH 3 ] group) and chromatin or histone modification. DNA methylation occurs in cytosines that precede guanines, known as dinucleotide CpGs or CpG islands, which occur in the promoter regions of many genes and generally results in gene silencing. However, changes in methylation may also occur in non-CpG-island regions, usually in close proximity to CpG islands. In cancer, methylation of cancer genes is more common than loss-of-function mutations or deletions .
Apoptosis
An equilibrium between proliferation and cell death is essential for normal homeostasis, and an imbalance can lead to abnormal growth. Cell death is important for tissue remodeling during embryogenesis and for maintaining homeostasis in normal adult tissues. Cell death is equally crucial for the removal of damaged cells, and this can occur via two different mechanisms – necrosis or apoptosis . Necrosis involves poor nutrient supply leading to membrane disruption and cell lysis without de novo protein synthesis.
In contrast, apoptosis is regulated and requires mRNA and protein synthesis. Microscopically, apoptosis is characterized by the appearance of apoptotic bodies composed of cell membrane remnants and condensed chromatin. Apoptosis-related proteases, termed caspases, become activated and affect signal transduction pathways by activation of: (1) enzyme precursors that digest genomic DNA into 200 base pair (bp) units; and (2) proteases that degrade structurally important proteins such as lamins and actin ( Fig. 107.4 ). Apoptosis can be induced by several different stimuli, including DNA damage (e.g. due to radiation, chemicals), withdrawal of growth cytokines (e.g. EGF, TGF-α, IGF, PDGF), and death-promoting agents (e.g. TNF) (see Fig. 107.4 ). Conflicts between different signaling pathways can also provoke apoptosis.
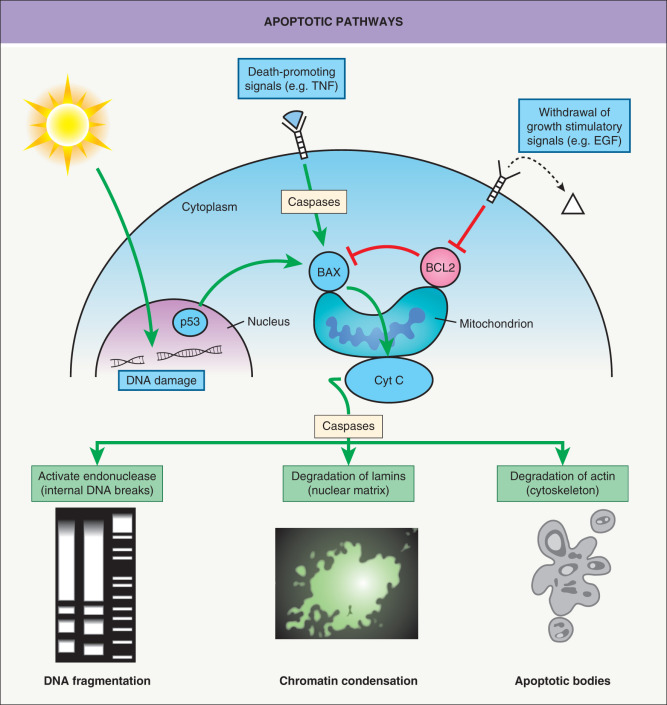
Independent of triggering apoptotic stimuli, apoptosis converges into a common series of molecular events that lead to irreversible morphologic changes and ultimately cell death. The regulation of apoptosis is under the strict control of proteins, including BCL-2 and BAX, both members of the BCL-2 family of proteins that are capable of regulatory cross-talk by heterodimerization of protein subunits. The mitochondrial membranes contain proteins that either activate apoptosis (BAX) or inhibit apoptosis (BCL-2). Overexpression of the survival factor BCL-2 blocks apoptosis and thus protects the cells against radiation and chemotherapeutic agents. The p53 protein activates transcription of BAX and inhibits transcription of BCL2 , the net result favoring apoptosis. Thus, the p53 protein is a key factor for integrating pathways regulating DNA synthesis, DNA repair, and apoptosis.
Limitless Replicative Potential
The number of mutations in a cell increases with time and partially explains the increasing risk for acquiring cancer as we age. The increased number of mutations is evident in normal cultured cells, which undergo ~60–70 doublings before reaching senescence and then death as a consequence of accumulating mutations and telomere shortening. Senescent cells are viable, but they are incapable of proliferation. During senescence, abnormal chromosomes accumulate and the telomere sequences diminish, eventually resulting in a crisis that leads to apoptosis.
The progressive erosion of telomere sequences (50–100 bp per mitosis) through successive cycles of replication eventually precludes protection of the ends of the chromosomes (see Fig. 67.22 ). This facilitates end-to-end chromosomal fusions, resulting in karyotypic disarray and then apoptosis. Carcinogenesis involves disruption of these normal apoptotic events, yielding limitless replicative potential for the tumor cells. Up to 90% of all cancers exhibit increased levels of telomerase, the enzyme required for maintenance of telomere sequences in normal cells; this leads to retention of telomeres irrespective of the number of cell divisions .
Cancer cell populations also contain a subpopulation of self-renewing cells called cancer stem cells. These latter cells can increase in number as tumors enlarge and they can give rise to progeny that have the ability to invade locally as well as metastasize to distant sites, referred to as tumor-propagating cells (TPCs) . Markers such as CD133, CD44 and ALDH (aldehyde dehydrogenase) are expressed by TPCs, and this population can be expanded within tumors by symmetrical division of TPCs and by cancer cells undergoing epithelial–mesenchymal transition.
Angiogenesis
Formation of new blood vessels (angiogenesis) is essential for tumor mass expansion, in order to provide adequate levels of oxygen and nutrients . Endothelial cells are normally quiescent, with low proliferative rates and mitoses rarely being observed (aside from the setting of wound healing). Angiogenesis is initiated by growth factors, e.g. fibroblast growth factor (FGF) and vascular endothelial growth factor (VEGF), acting in a paracrine fashion and stimulating local endothelial proliferation (see Ch. 102 ). Sprouts from surrounding vasculature migrate toward the tumor via protease-mediated remodeling of extracellular matrix (e.g. by serine proteases and metalloproteinases), thereby establishing new matrix–cell contacts. Of note, cancer cells express increased levels of VEGF and FGF. Inhibitors of angiogenesis, e.g. angiostatin, endostatin and thrombospondin, have been identified in plasma and extracellular matrix. Because thrombospondin is activated by p53, inactivation of p53 facilitates angiogenesis.
Invasion/Metastasis
Cell–cell interactions and extracellular glycoprotein matrix recognition are achieved by four classes of membrane-bound receptors, consisting of integrins, cadherins, selectins, and members of the immunoglobulin family. All of these cellular adhesion molecules have an extracellular domain that binds ligands, leading to a conformational change in the cytoplasmic tail of the receptor. Upon ligand binding, internal regions of the receptors bind to specific cytoplasmic proteins influencing various pathways involved in proliferation, cell migration, differentiation, and apoptosis. The extracellular ligands for integrins are matrix components composed of collagen, fibronectin or laminin (see Fig. 141.9 ) and immunoglobulin family members (see Table 102.3 ). Cadherins bind to cadherins present on adjacent cells and selectins bind to carbohydrate chains.
Disruption of tissue organization is a hallmark of cancer and is the result of alterations in cellular adhesion receptors, resulting in selective growth advantage. In cell cultures, dysregulation of cell adhesion pathways underlies anchorage-independent growth. Metastases from epithelial cancers often lose E-cadherin expression, facilitating the escape of cells from the primary site to other locations. E-cadherin expression can be inactivated by hypermethylation of regulatory regions as well as mutations in the coding sequence. Alternatively, inactivation occurs through proteolysis of the extracellular domain of E-cadherin or mutations in the gene encoding E-cadherin’s intracellular ligand, β-catenin.
Tumors are not comprised purely of neoplastic cells. Rather, they share properties with organs, including being composed of multiple cell types, possessing an extracellular matrix, and having both blood and lymphatic vessels . Signaling between neoplastic cells and the tumor constituent cells, e.g. fibroblasts, macrophages and endothelial cells, appears to be important for the growth of a tumor and is often induced by signals from the neoplastic cells . Reciprocal release of abundant growth-stimulating signals creates amplifying loops enabling growth of both the tumor and its cognate stroma.
In humans, the most common cancers are basal cell carcinoma (BCC) and squamous cell carcinoma (SCC), often termed “non-melanoma skin cancers”. Although BCCs and SCCs possess many similar features, there are fundamental biologic differences, justifying a distinction between the two and rendering “non-melanoma skin cancer” a poor conceptual term. In the remainder of this chapter, BCC and SCC will be described with an emphasis on the similarities and the differences between their tumor biology. Disruption of the Hedgehog–Patched signaling pathway is closely linked to development of BCC while the TP53 gene is often mutated in both BCC and SCC. The Patched (PTCH1) and TP53 genes will serve as a model to illustrate some of the basic features of epithelial skin carcinogenesis.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


