Abstract
A diverse array of entities can present with cutaneous vasculitis, and the characteristic skin findings reflect the caliber of inflamed blood vessel – small, medium-sized, or large. While cutaneous small vessel vasculitis is often idiopathic, it may be due to an infection (e.g. hepatitis C virus), medication (e.g. penicillins), autoimmune connective tissue disease (e.g. Sjögren syndrome), or systemic vasculitis (e.g. granulomatosis with polyangiitis [Wegener granulomatosis]). As a result, after the diagnosis of cutaneous vasculitis has been established via clinicopathologic correlation, further evaluation includes a directed search for any underlying etiology and evidence of systemic involvement. Treatment of cutaneous vasculitis depends upon many factors including the underlying cause, severity, and sites of internal involvement.
Keywords
ANCA-associated vasculitis, cryoglobulin, cutaneous small vessel vasculitis, cutaneous vasculitis, erythema elevatum diutinum, Henoch–Schönlein purpura, IgA vasculitis, leukocytoclastic vasculitis, purpura, urticarial vasculitis, acute hemorrhagic edema of infancy, cryoglobulinemic vasculitis, granulomatosis with polyangiitis, Wegener granulomatosis, eosinophilic granulomatosis with polyangiitis, Churg–Strauss syndrome, microscopic polyangiitis, polyarteritis nodosa, temporal arteritis
- ▪
Cutaneous signs of vasculitis are a reflection of the size of the vessels involved
- ▪
Vasculitis can be limited to the small vessels of the skin or it can be a sign of life-threatening internal organ involvement
- ▪
The clinical diagnosis of cutaneous vasculitis requires histopathologic confirmation, and multiple biopsies may be required
Introduction
Vasculitis represents a specific pattern of inflammation of the blood vessel wall and it can occur in any organ system of the body. Cutaneous vasculitis may be: (1) a skin-limited disease; (2) a primary cutaneous vasculitis with secondary systemic involvement; or (3) a cutaneous manifestation of a systemic vasculitis.
Vasculitis can affect small, medium-sized, or large vessels of the arterial and/or venous systems ( Table 24.1 ). Small vessels include arterioles, capillaries, and postcapillary venules, which are found in the superficial and mid dermis of the skin. Medium-sized vessels refer to the small arteries and veins that reside within the deep dermis or subcutis. Large vessels include the aorta and named arteries. Cutaneous involvement occurs almost exclusively with vasculitis of small and medium-sized vessels; therefore, the large vessel vasculitides are discussed briefly or mentioned in Table 24.1 .
| CUTANEOUS VASCULITIS CLASSIFICATION SCHEME | |||
|---|---|---|---|
| Caliber of the predominantly affected vessel | Classification | Subclassification or etiologies | Morphology of cutaneous lesions |
| Small | Cutaneous small vessel vasculitis (CSVV) |
|
|
| Small and medium-sized (“mixed”) | ANCA-associated |
|
|
| Secondary causes |
| ||
| Medium-sized | Polyarteritis nodosa (PAN) |
|
|
| Large * | Temporal arteritis |
| |
| Takayasu arteritis (disease) |
| ||
In this chapter, cutaneous vasculitis refers to vasculitis with any underlying etiology and affecting any sized vessel in which the clinical manifestations include the skin. Cutaneous small vessel vasculitis (CSVV) is synonymous with cutaneous leukocytoclastic vasculitis (LCV) and refers to involvement of the postcapillary venules of the dermis by intense neutrophilic vascular inflammation.
Classification
Two major classification schemes are the American College of Rheumatology 1990 criteria and the 2012 revised International Chapel Hill Consensus Conference Nomenclature system . Because a system based upon the predominant size of the involved blood vessel helps to predict the clinical presentation and the corresponding histologic findings, this type of classification scheme will be employed in this chapter (see Table 24.1 ). Additional features that aid in classifying cutaneous vasculitis are systemic manifestations, direct immunofluorescence findings, and the presence or absence of antineutrophil cytoplasmic antibodies (ANCAs). Despite descriptions of distinct vasculitic syndromes, there are no unique diagnostic criteria, and clinicopathologic correlation is always required.
Epidemiology
The annual, population-based incidence of biopsy-proven cutaneous LCV is ~45 per million; this figure includes various subtypes such as urticarial vasculitis, cryoglobulinemic vasculitis, and ANCA-associated vasculitis . Cutaneous vasculitis occurs in all age groups (mean age in adults, 47 years; mean age in children, 7 years), has a slight female predominance , and is much more common in adults than in children. The majority of children have Henoch–Schönlein purpura.
Pathogenesis
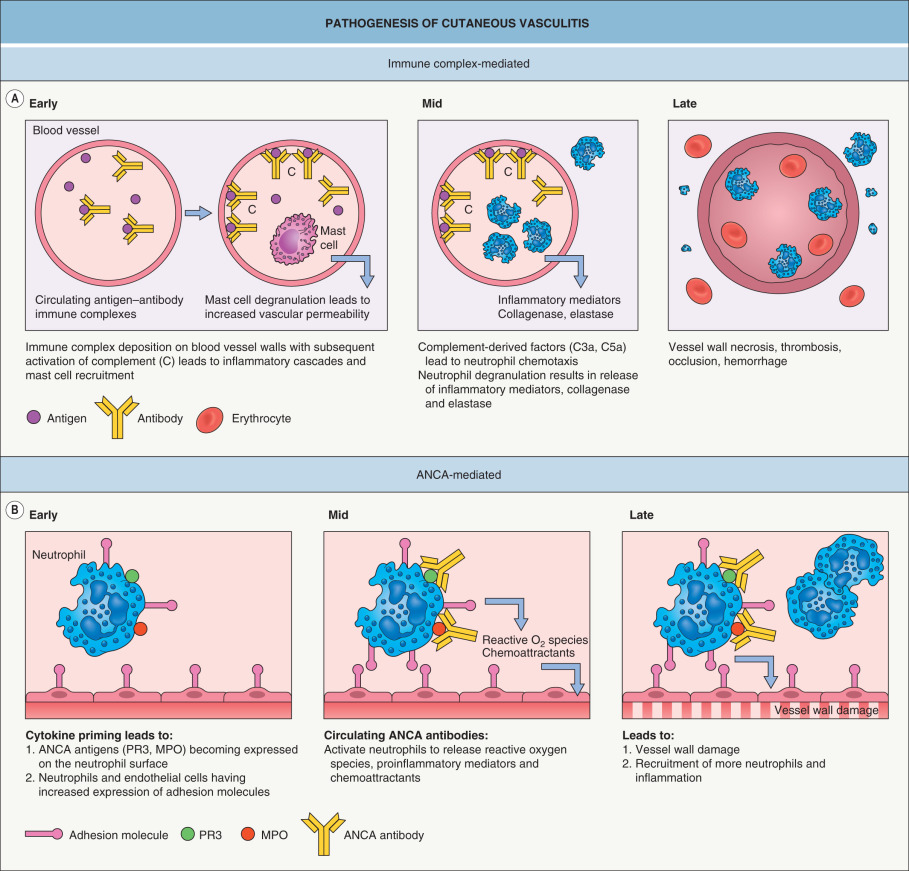
CSVV is mediated by immune complexes that form in the presence of antigen excess, and after their deposition within postcapillary venules, these complexes lead to complement-mediated chemotaxis of neutrophils ( Fig. 24.1A ). In ANCA-associated vasculitides, vessel wall damage is directly mediated by neutrophils rather than by immune complex deposition ( Fig. 24.1B ), hence the term “pauci-immune” vasculitides.
General Clinical Features
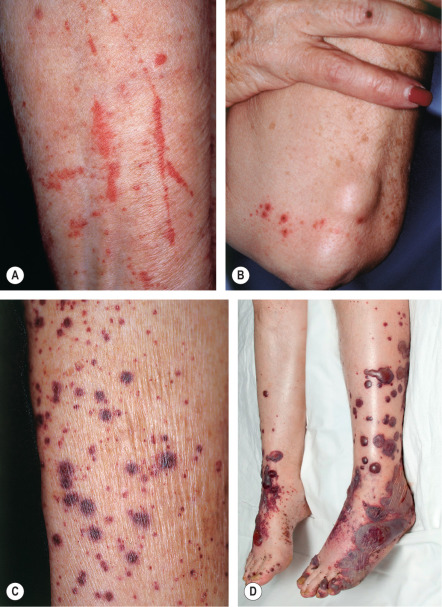
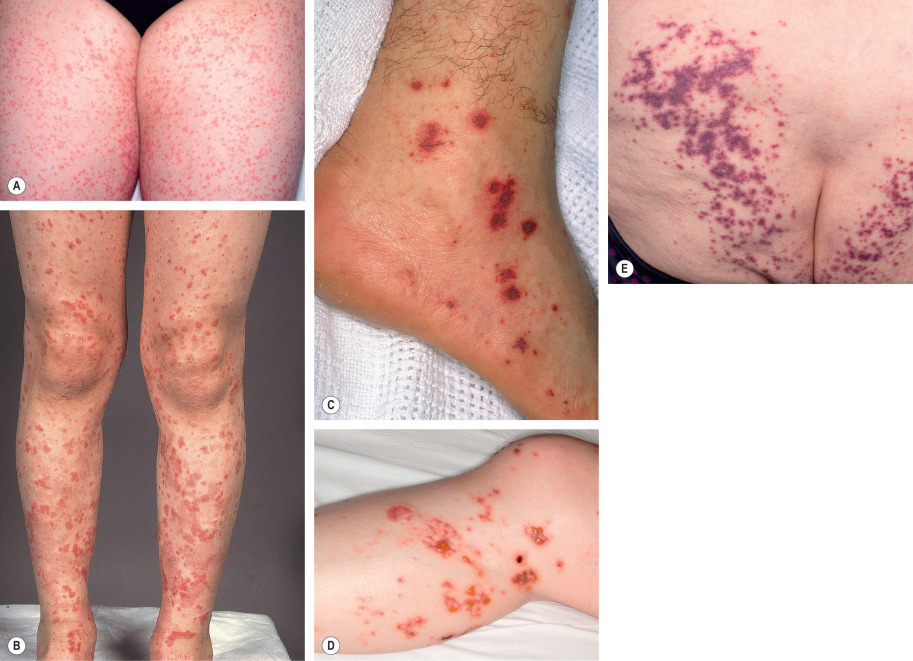
The skin lesions of CSVV usually appear 7–10 days after the triggering event. In systemic vasculitic syndromes, signs of systemic involvement often precede the appearance of associated cutaneous lesions (average, 6 months), but the interval can be as short as days or as long as years . As noted previously, the cutaneous findings of vasculitis depend upon the predominant size of the vessels that are involved. CSVV typically presents with palpable or macular purpura, but urticarial papules, pustules, vesicles, petechiae, or targetoid lesions can be seen ( Figs 24.2 & 24.3 ). The lesions favor dependent sites, as well as areas under tight-fitting clothing, reflecting the influence of hydrostatic pressure and stasis on the pathophysiology. In general, the lesions are asymptomatic, but they may itch, burn, or sting.
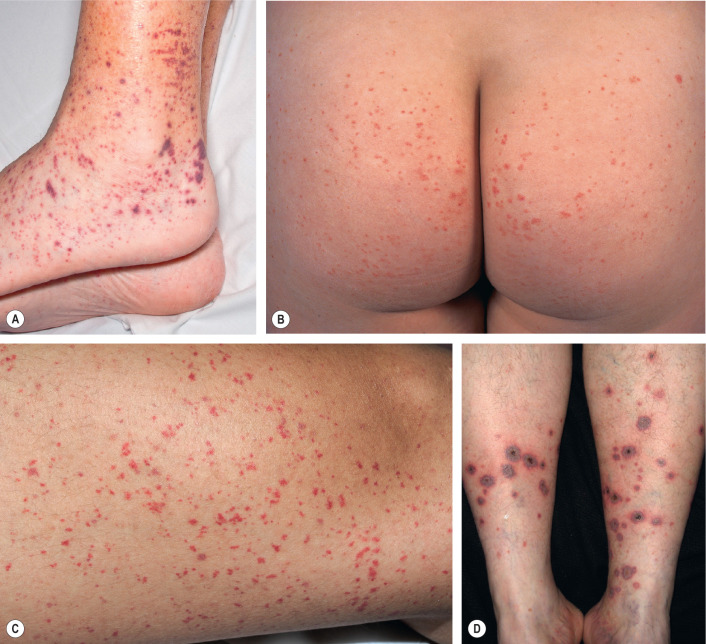
In medium-sized vessel vasculitis, the affected blood vessels reside within the reticular dermis or subcutis. As a result, the latter typically presents with livedo racemosa, retiform purpura, ulcers, subcutaneous nodules, and/or digital necrosis. In general, the presence of ulcers or necrosis suggests deeper arterial involvement. The combination of palpable purpura (or other signs of CSVV) plus signs of medium-sized vessel disease points to a “mixed” pattern of vasculitis (see Table 24.1 ), as is seen in ANCA-associated vasculitides or autoimmune connective tissue disease-associated vasculitis.
Arthralgias and arthritis as well as constitutional symptoms such as fever, weight loss, and malaise can be manifestations of vasculitis of any sized vessel . For patients with systemic involvement, presenting symptoms and signs (e.g. abdominal pain, paresthesias, hematuria) will vary according to the affected organs. In a population-based study of 84 patients with biopsy-proven cutaneous LCV, 39 patients (46%) had systemic manifestations, with renal involvement most commonly observed (17 of 39 patients); recurrent disease (mean duration of disease activity, 2 years) was observed in 30% of patients .
Pathology
Timing and appropriate sampling of the most likely involved vessel increase the diagnostic yield of a skin biopsy. Ideally, lesions should be biopsied within the first 24 to 48 hours of appearance. When CSVV is suspected, a biopsy specimen for direct immunofluorescence should be obtained, as the presence of certain immunoglobulins (e.g. IgA) may suggest a particular diagnosis and influence prognosis. In general, punch biopsies are adequate for the diagnosis of CSVV while an incisional or excisional biopsy may be required to diagnose vasculitis of larger vessels.
In cutaneous vasculitis, the histologic findings vary depending upon the type and age of the sampled lesion in addition to the size of the affected vessel. The classic histopathologic features of CSVV are referred to as LCV and consist of transmural infiltration of the walls of small vessels (primarily postcapillary venules) by neutrophils undergoing karyorrhexis of their nuclei, as well as fibrinoid necrosis of the damaged vessel walls ( Fig. 24.4 ). Other findings include leukocytoclasia (degranulation and fragmentation of neutrophils, leading to the production of nuclear dust), extravasated erythrocytes, and signs of endothelial cell damage. However, lesions present for greater than 48 to 72 hours may have a predominantly mononuclear rather than neutrophilic infiltrate. Palpable purpura, the most common clinical lesion of CSVV, can be explained by the infiltrate of leukocytes (palpability) and the resulting extravasation of RBCs from the damaged blood vessel (purpura).
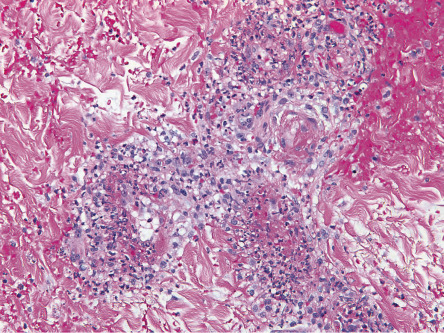
Additional histological findings may provide clues to the underlying etiology, e.g. eosinophils in drug-induced CSVV (up to 100% of cases) , thrombi plus CSVV in patients exposed to levamisole-tainted cocaine, and thrombi plus dense dermal inflammation in septic vasculitis. Involvement of deeper dermal vessels alerts the clinician to look for an underlying etiology rather than presuming the CSVV is idiopathic .
Vasculitis of medium-sized blood vessels is characterized by similar changes involving the vessels (e.g. small arteries) of the deep reticular dermis and subcutaneous fat. Neovascularization of the adventitia, in the form of small capillaries, is commonly seen in older lesions of medium-sized vessel vasculitis.
In ~80% of cases of CSVV, direct immunofluorescence (DIF) demonstrates deposition of C3, IgM, IgA and/or IgG (generally in that order of frequency) in a granular pattern within the vessel walls . Immunoglobulin deposition is highest (up to 100%) in skin lesions present for ≤48 hours . On the other hand, in 30% of samples obtained 48–72 hours after lesion onset, DIF will be negative for immunoglobulins, and only C3 will be detected in lesions present for >72 hours . In patients with an ANCA-positive vasculitis, the DIF of lesional skin is usually negative. After controlling for duration, it is preferable to biopsy more proximal lesions for DIF in order to avoid nonspecific vascular fluorescence that can occur in sites of greatest hydrostatic pressure.
As stated previously, the term CSVV is routinely equated with LCV. However, there are additional forms of cutaneous vasculitis, in particular lymphocytic ( Table 24.2 ) and granulomatous (see below) . Use of the term lymphocytic vasculitis often requires clarification, especially in discussions with non-dermatologists.
| LYMPHOCYTIC VASCULITIS |
|---|
|
Differential Diagnosis
Several disorders can present with skin lesions that resemble the various cutaneous manifestations of vasculitis and they are listed in Table 24.3 .
| CLINICAL DIFFERENTIAL DIAGNOSIS OF CUTANEOUS VASCULITIS | |
|---|---|
| Clinical presentation | Differential diagnosis |
| Palpable purpuric papules/plaques |
|
| Purpuric macules/patches |
|
| Urticarial lesions |
|
| Ulcers ± atrophie blanche |
|
| Nodules |
|
| Livedo reticularis | See Chapter 106 |
| Digital gangrene |
|
Cutaneous Small Vessel Vasculitis
▪ Cutaneous leukocytoclastic vasculitis ▪ Cutaneous leukocytoclastic angiitis ▪ Hypersensitivity angiitis ▪ Cutaneous necrotizing venulitis
- ▪
Palpable purpura, urticarial lesions, and/or hemorrhagic macules or vesicles; occasionally, targetoid lesions, pustules, and ulcerations
- ▪
Lesions favor the lower extremities (especially the ankles), dependent areas, or pressure points
- ▪
Only involves small vessels, primarily postcapillary venules
- ▪
Histopathologically, leukocytoclastic vasculitis is seen
- ▪
Extracutaneous involvement occurs in up to 30% of patients, but it is usually mild
Introduction
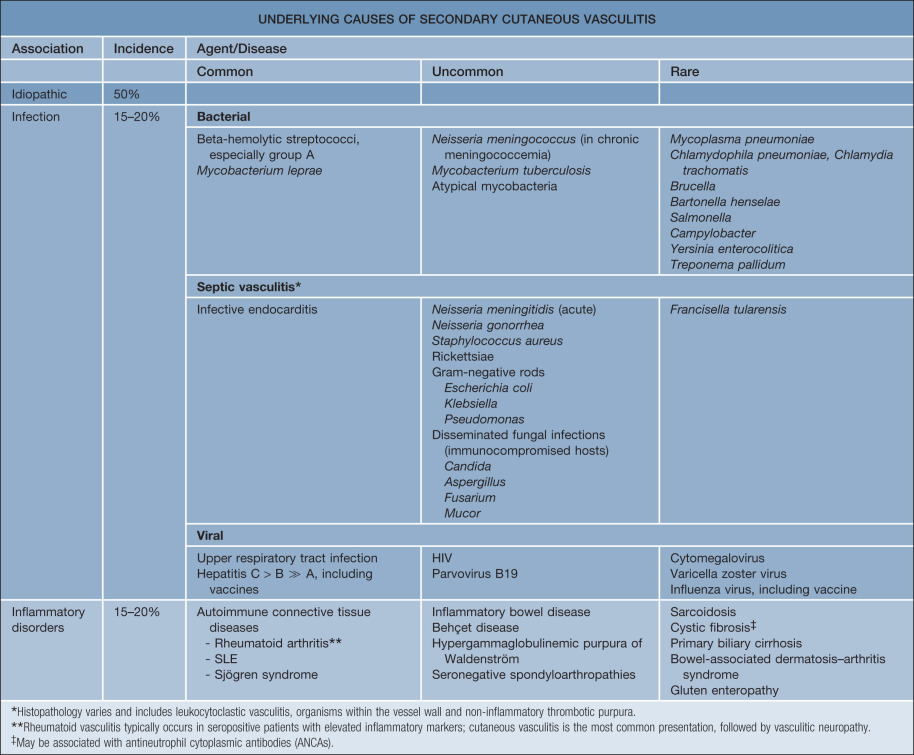
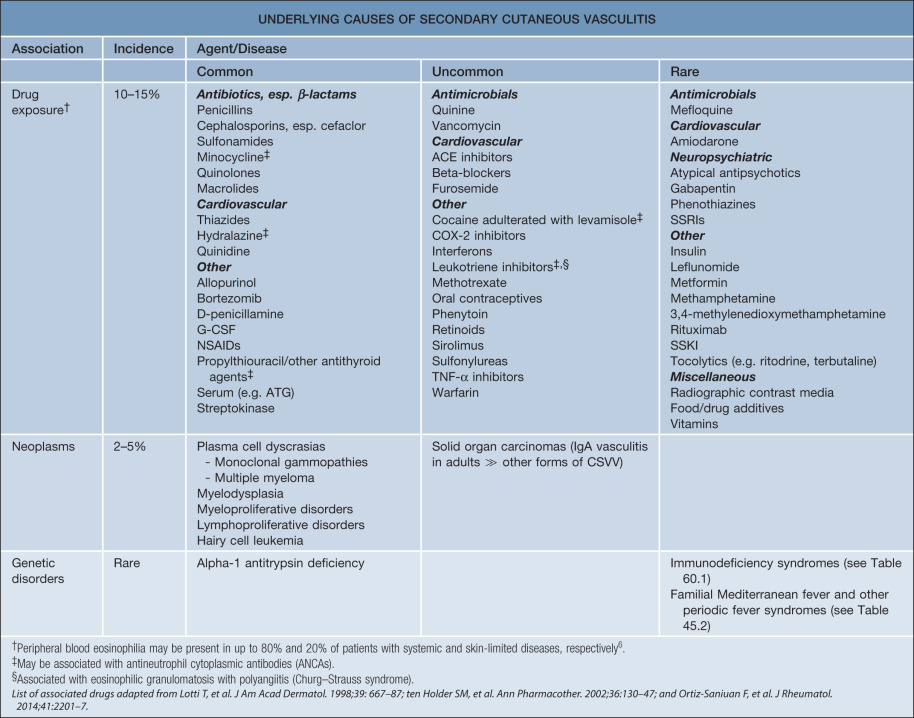
CSVV is a vasculitic process that involves primarily the dermal postcapillary venules and is characterized histologically by LCV. Although CSVV with LCV can be seen in the setting of mixed (small and medium-sized vessel) vasculitides (see Table 24.1 ), the term CSVV is generally reserved for small vessel vasculitis of the skin without medium-sized vessel involvement, irrespective of the clinical severity of the skin disease or the underlying etiology. CSVV is often idiopathic in nature, but may be secondary to an underlying cause such as an infection or medication ( Table 24.4 ). Within the spectrum of CSVV are several subtypes whose rather unique epidemiologic and clinical features warrant subclassification (see Table 24.1 ) and separate discussion.
Epidemiology
CSVV occurs in both sexes and at all ages , but is more common in the adult population. It is estimated that ~10% of those affected are children. The annual population-based incidence of CSVV (excluding those with a distinct clinical subtype of CSVV) is 21 cases per million .
Pathogenesis
CSVV is mediated by immune complex deposition (see Fig. 24.1A ).
Clinical features
CSVV typically presents after exposure to an inciting agent with a single crop of lesions consisting of palpable purpura, erythematous papules, urticarial lesions, or hemorrhagic vesicles that range in size from 1 mm to several centimeters (see Figs 24.2 & 24.3 ). Lesions often begin as a purpuric macule or partially blanching urticarial papule. Occasionally, pustules, ulcerations, and targetoid lesions are seen. CSVV favors dependent areas, as well as areas affected by trauma (Koebner phenomenon) or under tight-fitting clothing. Even exercise, in particular walking or hiking in hot weather, can induce CSVV on the lower extremities. Although they are usually asymptomatic, the lesions can be associated with burning, pain, or pruritus. Residual postinflammatory hyperpigmentation may persist for months after the primary process resolves.
Constitutional symptoms, such as fevers, weight loss and myalgias, may accompany flares of CSVV. Systemic symptoms develop in 5–25% of patients with CSVV, with arthralgias and arthritis occurring most commonly (15–65%), followed by genitourinary (3–7%) signs or symptoms, and gastrointestinal involvement (3–5%) . A recent population-based study demonstrated systemic involvement in 11 of 38 patients (29%) with CSVV , which is a lower figure than was cited previously because those individuals with a distinct clinical subtype of CSVV were excluded. In general, signs or symptoms of gastrointestinal, renal, or neurologic involvement should increase the clinical suspicion for a systemic vasculitis. In one study, the presence of paresthesias or fever and the absence of painful lesions were identified as risk factors for an associated systemic disease .
While the prognosis of patients with CSVV depends upon the severity of systemic involvement, approximately 90% of patients will have spontaneous resolution of cutaneous lesions within several weeks or a few months, while another 10% will have chronic or recurrent disease at intervals of months to years . In the latter group, the average duration of disease activity was 24–28 months . The presence of arthralgias or cryoglobulinemia and an absence of fever may portend chronicity . An underlying cause for the CSVV, such as an autoimmune connective tissue disease or neoplasm, will also affect prognosis.
Differential diagnosis
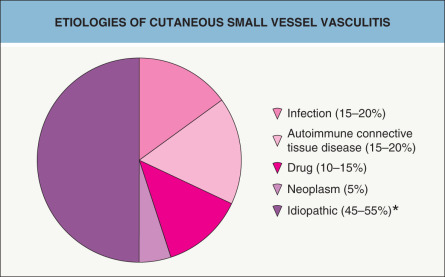
In addition to determining if the patient has a particular subtype of CSVV (see Table 24.1 ) or has CSVV as a manifestation of a systemic vasculitic syndrome, causes of secondary cutaneous vasculitis also need to be considered (see Table 24.4 ; Figs 24.5 & 24.6 ). Entities listed in Table 24.3 , especially the first three categories, need to be included in the clinical differential diagnosis.
Treatment
The initial intervention in CSVV consists of supportive care and identifying possible triggers. The need for and choice of additional treatments depends on the severity of the cutaneous involvement, chronicity, and whether or not systemic involvement is present. Specific therapeutic options are outlined in Table 24.10 .
CSVV often resolves without any treatment other than avoidance of the inciting trigger. For mild skin-limited disease, supportive measures (e.g. leg elevation, avoiding tight clothing, rest, compression stockings) or symptomatic therapy (e.g. antihistamines, NSAIDs) may be all that is necessary.
Chronic (>4 weeks’ duration) or more severe cutaneous disease may require more aggressive systemic therapy. Colchicine and dapsone may be used either alone or in combination. Oral colchicine (0.6 mg two to three times daily) is helpful for both skin and joint manifestations. However, gastrointestinal side effects are fairly common, even at low doses. Oral dapsone (50–200 mg/day) can lead to improvement of mild to moderate chronic lesions.
Patients with severe, ulcerating, or progressive cutaneous disease who require rapid control of symptoms can be treated with a short course of high-dose oral corticosteroids (e.g. up to 1 mg/kg/day of prednisone). Because of the multiple adverse effects of long-term oral corticosteroids, a taper over 4 to 6 weeks should be attempted. If the patient develops recurrent CSVV as the dosage is decreased, addition of a steroid-sparing agent is warranted. Immunosuppressive agents such as azathioprine (2 mg/kg/day) and methotrexate (<25 mg weekly) have been reported to be useful in recalcitrant CSVV.
Henoch–Schönlein Purpura (and IgA Vasculitis in Adults)
▪ Schönlein–Henoch purpura ▪ Anaphylactoid purpura ▪ Purpura rheumatica ▪ Cutaneous small vessel vasculitis secondary to IgA immune complexes
- ▪
Most commonly occurs in children <10 years of age and in association with a preceding respiratory infection, but may also be seen in adults
- ▪
Intermittent palpable purpura on extensor extremities and buttocks
- ▪
IgA-dominant immune deposits in walls of small blood vessels
- ▪
Arthralgias and arthritis
- ▪
Abdominal pain and/or melena
- ▪
Renal vasculitis often mild but can be chronic
- ▪
In adults, may be associated with an underlying malignancy
Introduction
Henoch–Schönlein purpura (HSP) is a specific form of CSVV with vascular IgA deposition that typically affects children following a respiratory tract infection, but may also occur in adults. The classic tetrad consists of palpable purpura, arthritis, abdominal pain, and hematuria. Although the relationship between HSP and other forms of vasculitis in which vascular IgA deposition is present remains controversial, the presence of IgA deposition within the walls of small blood vessels has clinical and prognostic implications in both children and adults.
Epidemiology
HSP is the most common form of vasculitis in children, with an incidence of 30 to 270 cases per million children per year . The average age of onset is 6 years, and 90% of cases occur in children <10 years of age . In adults, the incidence of HSP is 8 to 18 cases per million , and the population-based incidence of biopsy-proven cutaneous IgA vasculitis is 13 cases per million per year . The disorder follows a seasonal pattern, with a peak occurrence during the winter. HSP has a slight male predominance in both children and adults.
Pathogenesis
HSP frequently presents 1 to 2 weeks following an upper respiratory tract infection, especially in children. Although several studies have reported that 20–50% of HSP patients have positive antistreptolysin O titers , no causal role for group A β-hemolytic streptococci has been demonstrated.
IgA (specifically IgA1) is thought to play a fundamental role in the pathogenesis of HSP, as IgA deposits in the postcapillary venules of the skin and mesangium and, less commonly, circulating IgA-containing immune complexes and increased serum levels of IgA, have been demonstrated in patients with HSP . In a recent case series of over 400 HSP patients, increased serum IgA levels were found in ~30% . Lack of glycosylation of the hinge region of IgA1 may promote the formation of macromolecular complexes that lodge within the mesangium and activate the alternate complement pathway .
Certain genetic polymorphisms may predispose to more severe disease in HSP . For example, HLA-B35 positivity may predispose to renal disease, while patients who do not have the ICAM-1 469 K/E variant have less severe gastrointestinal involvement .
Clinical features
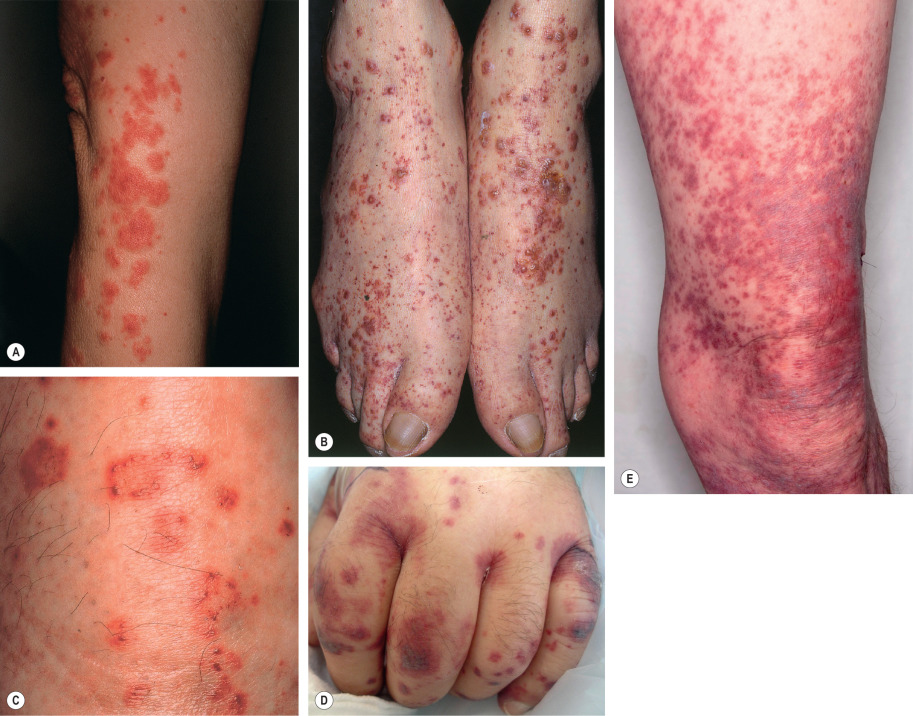
HSP classically presents with purpura (100% of patients) in association with arthritis, abdominal pain, and/or nephritis . Fever occurs in approximately 20% of adults and 40% of children . Cutaneous lesions begin as erythematous macules or urticarial papules and evolve into palpable purpura ranging in size from pinpoint to several millimeters ( Fig. 24.7 ). Urticaria, vesicles, bullae, targetoid lesions, and foci of necrosis can also be seen. Typically, lesions are symmetrically distributed on the buttocks and lower extremities, but may also involve the trunk, upper extremities, and face. Individual lesions usually regress within 10 to 14 days, with resolution of cutaneous involvement over a period of several weeks to months. Recurrences of skin disease are observed in 5–10% of patients .
Extracutaneous manifestations of HSP are common. Arthritis occurs in up to 75% of patients with HSP and most commonly affects the joints of the lower extremities (knees and ankles). Gastrointestinal involvement (50–75% of patients) may precede the purpura and presents with colicky abdominal pain (65%), gastrointestinal bleeding (30%), and/or vomiting. Intussusception and bowel perforation are rare complications.
Renal involvement occurs in 40–50% of patients and typically presents with microscopic hematuria (40%), often accompanied by proteinuria (25%). Although the appearance of cutaneous lesions often precedes the development of nephritis, the latter is clinically evident within 3 months . In pediatric patients, risk factors for the development of nephritis include age >8 years at onset, abdominal pain, and recurrent disease . Depending upon the series, persistent renal disease has been observed in 8–50% of patients , emphasizing the need for longitudinal monitoring until all abnormalities resolve. Fortunately, only 1–3% of children develop long-term renal impairment . Poor prognostic factors include renal failure at the time of onset, nephrotic syndrome, hypertension, and decreased factor XIII activity .
In young boys, orchitis is a rare manifestation of systemic disease. The lung is also a rare site of involvement, presenting as hemoptysis and/or pulmonary infiltrates due to diffuse alveolar hemorrhage .
IgA small vessel vasculitis in adults, termed adult HSP by some authors, should be considered separately, as the clinical presentation and prognosis differ from that in children. For example, necrotic skin lesions are present in 60% of adults while cutaneous necrosis is observed in <5% of children . Adults with IgA vasculitis are also more likely than children to develop chronic renal insufficiency (up to 30%) , especially if they have fever and an elevated ESR . There are conflicting data as to whether the risk of renal insufficiency is increased if purpura are present above the waist . When CSVV is due to an underlying neoplasm, the latter is most often a hematologic malignancy rather than a solid organ malignancy (see Table 121.6 ). In contrast, 60–90% of adult patients with neoplasm-associated IgA vasculitis will have cancer of a solid organ, in particular the lung . Adults are also more likely than children to have diarrhea and leukocytosis, to require more aggressive therapy, and to have a longer hospital stay .
Pathology
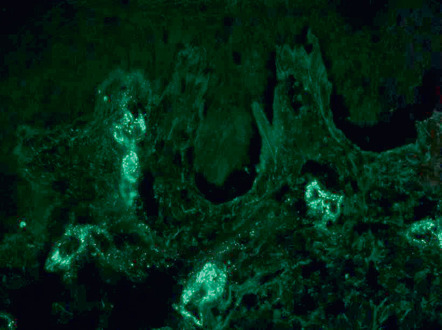
Leukocytoclastic vasculitis of the small dermal blood vessels is seen. In one series of adults with IgA vasculitis (age >40 years), an absence of eosinophils nearly tripled the risk of renal involvement . DIF demonstrates perivascular IgA and C3 deposits ( Fig. 24.8 ) . Other immunoglobulins (IgM, IgG) may be seen, but are generally not as prominent as IgA and are not associated with renal involvement . Perivascular C3 deposits and papillary dermal edema were associated with renal involvement in one series of pediatric patients . In a study of adults with glomerulonephritis who had IgA deposition in both the skin and the kidneys, concordance of DIF for other immunoconjugates (in biopsies from these two tissues) ranged from 50–80% . Of note, a small subset of patients has been described that meets clinical criteria of HSP but lacks IgA deposition on DIF . By indirect immunofluorescence, IgA ANCAs may be present in some patients.
Differential diagnosis
The clinical differential diagnosis includes any of the vasculitides involving small vessels (see Table 24.1 ) as well as several of the entities in Table 24.3 . Because up to 80% of all adults with CSVV may demonstrate some vascular IgA deposition and IgA deposition can be seen in other diseases (e.g. drug hypersensitivity, IgA monoclonal gammopathy, inflammatory bowel disease, lupus erythematosus, cryoglobulinemia) , a diagnosis of HSP is supported by IgA predominance in the correct clinical setting.
Treatment
Because HSP is generally self-limited and resolves over the course of weeks to months, treatment is mainly supportive. Dapsone and colchicine may decrease the duration of cutaneous lesions and frequency of recurrences (see Table 24.10 ) . Systemic corticosteroids are effective in treating the arthritis and abdominal pain associated with HSP, as well as reducing the gastrointestinal complications and duration of skin lesions, but do not prevent recurrences of purpura . Referral to a nephrologist is appropriate for patients with evidence of renal involvement. In adults, the following factors may predict relapsing disease: age >30 years, an underlying systemic disorder, persistent purpura >1 month, abdominal pain, hematuria, and absence of IgM on DIF .
Considerable controversy surrounds the use of corticosteroids and/or immunosuppressive medications for the treatment of severe renal disease and for preventing renal sequelae in individuals who have severe renal involvement. In one study, the use of prednisone was associated with a more rapid resolution of renal disease during the 4-week treatment period . However, a Cochrane review of interventions for preventing and treating the renal disease of HSP found no difference in the risk of persistent kidney disease (at 6–12 months) in those children treated with prednisone (2–4 weeks) at the time of presentation as compared to placebo or supportive therapy . This review also found no difference in the risk of persistent renal disease in patients with severe kidney disease who were treated with cyclophosphamide versus supportive care. In a recent randomized, double-blind, placebo-controlled trial involving over 300 children with HSP, early treatment with 14 days of prednisolone did not reduce the prevalence of proteinuria 12 months after disease onset . Lastly, a meta-analysis that examined the use of systemic corticosteroids at the time of diagnosis versus supportive care found a decrease in the mean (but not the median) time to resolution of abdominal pain, while the odds of developing persistent renal disease were decreased . In sum, the current consensus appears to be that corticosteroids do not prevent renal disease but could be used to treat severe nephritis .
Acute Hemorrhagic Edema of Infancy
▪ Acute hemorrhagic edema of childhood ▪ Infantile acute hemorrhagic edema ▪ Finkelstein disease ▪ Seidlmayer syndrome ▪ Finkelstein–Seidlmayer disease ▪ Purpura en cocarde avec edema ▪ Postinfectious cockade purpura
- ▪
The child is well-appearing
- ▪
Seen primarily in children between 4 and 24 months of age
- ▪
Annular, circular, or targetoid purpuric plaques on the face and extremities
- ▪
Tender, non-pitting edema of acral sites
- ▪
Extracutaneous involvement rare
- ▪
Benign clinical course with spontaneous resolution within 1 to 3 weeks
Introduction
Acute hemorrhagic edema of infancy (AHEI) is an unusual form of CSVV that usually affects children 4 to 24 months of age. Cutaneous lesions typically begin as plaques with variable degrees of hemorrhage and favor the head and extremities. They subsequently become edematous and targetoid in appearance. Previously considered a benign form of HSP, AHEI is now recognized as its own clinical entity.
Epidemiology
AHEI is a relatively rare disorder. It affects children <2 years of age, with ~70% of cases occurring in boys . A seasonal variation is observed, with a peak incidence during the winter months.
Pathogenesis
Although the etiology of AHEI is unknown, 75% of patients have an associated infection, drug exposure, or immunization . An infectious prodrome is observed in two-thirds of patients, most commonly a respiratory tract infection (80%), diarrheal illness (12%), or urinary tract infection (6%) . There are multiple reported triggers and they are listed in Table 24.5 . As with other forms of CSVV, the pathogenesis of AHEI is thought to involve immune complex deposition in response to an antigenic trigger .
| TRIGGERS AND ASSOCIATIONS IN ACUTE HEMORRHAGIC EDEMA OF INFANCY AND URTICARIAL VASCULITIS |
|---|
| Acute hemorrhagic edema of infancy – triggers |
|
|
| Urticarial vasculitis – associations |
| Autoimmune connective tissue diseases (Sjögren syndrome, SLE) |
| Serum sickness |
| Cryoglobulinemia |
|
|
|
|
Clinical features
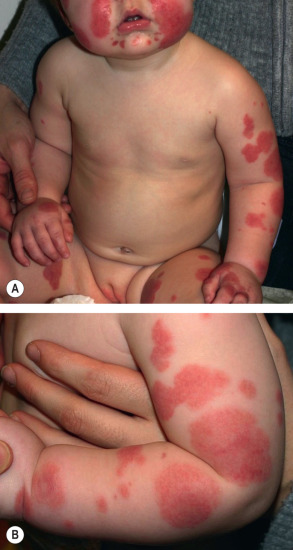
Children typically do not appear ill, although fever is present in ~45% of patients. The typical interval between the inciting event (see above) and the onset of disease is 1–2 weeks . Cutaneous involvement presents abruptly with large erythematous patches or urticarial plaques that then evolve into medallion, annular, iris, or targetoid purpuric plaques. The latter favor the cheeks, ears, and extremities ( Fig. 24.9 ), but truncal involvement may also occur. Lesions may be asymptomatic, painful, or, less commonly, pruritic. Arcuate, polycyclic, scalloped, or rosette-shaped lesions and vesicobullae occur less commonly, as does healing with atrophic scars . Tender, non-pitting edema of the face, ears, extremities (including the hands and feet), and scrotum is characteristic. Although mucosal and visceral involvement is rare, oral petechiae, conjunctival injection, abdominal pain, arthralgias, glomerulonephritis, and intussusception (<1%) may occur . The course is benign, with spontaneous and complete resolution without sequelae within 1 to 3 weeks. Although exacerbations may occur, once a remission has lasted 2 weeks, relapses as a rule are not observed .
Pathology
Leukocytoclastic vasculitis involving the capillaries and postcapillary venules of the upper and mid dermis is observed. By DIF, IgA deposits in a vascular pattern are seen in one-quarter to one-third of cases .
Differential diagnosis
Patients with features of both AHEI and HSP have been reported . An age of onset <2 years, disease confined to the skin, and a briefer duration are characteristic of AHEI. In young children with urticarial lesions and acrofacial edema, urticaria multiforme should be considered . The differential diagnosis also includes erythema multiforme, urticaria, urticarial drug eruptions, serum sickness-like reactions, Kawasaki disease, urticarial vasculitis, and Sweet syndrome. Given the significant cutaneous hemorrhage, trauma, acute meningococcemia, and purpura fulminans are sometimes considered. Routine laboratory tests are nonspecific, and the diagnosis is based on appropriate clinicopathologic correlation.
Treatment
Treatment is supportive (see Table 24.10 ). Antibiotics should be given to treat concurrent bacterial infections. Antihistamines may be useful for symptomatic relief. Systemic corticosteroids have not been shown to alter the course of the disease .
Urticarial Vasculitis
▪ Chronic urticaria as a manifestation of venulitis ▪ Urticaria and arthralgia with necrotizing angiitis
If hypocomplementemia: ▪ Hypocomplementemic vasculitis ▪ Hypocomplementemia with cutaneous vasculitis and arthritis
- ▪
Recurrent episodes of painful, persistent urticarial lesions that last >24 hours and often resolve with residual hyperpigmentation
- ▪
Angioedema may also be present
- ▪
May be associated with constitutional symptoms and arthritis
- ▪
Patients with hypocomplementemia are more likely to have systemic involvement
- ▪
Associated disorders include autoimmune connective tissue diseases (especially systemic lupus erythematosus, Sjögren syndrome) and viral infections
Introduction
Urticarial vasculitis is a clinicopathologic entity consisting of persistent urticarial lesions that demonstrate the histopathologic features of LCV. Its association with autoimmune connective tissue diseases and potential overlap with systemic lupus erythematosus (SLE) distinguishes this disorder from typical CSVV. Although the relationship between urticarial vasculitis and neutrophilic urticaria is an area of debate, in this chapter, the definition of urticarial vasculitis is as follows: urticarial lesions that histopathologically demonstrate vasculitis as defined by a minimum of leukocytoclasia with vessel wall necrosis , but with or without fibrinoid deposits, perivascular inflammation, or red blood cell extravasation .
Epidemiology
The population-based incidence of urticarial vasculitis is 5 cases per million per year . In patients with chronic urticaria in whom a strict definition of urticarial vasculitis (see above) is applied, the prevalence of urticarial vasculitis is ~5% , although this may be an overestimation. The peak incidence is during the fifth decade, and 60–80% of patients with urticarial vasculitis are female. The hypocomplementemic form occurs almost exclusively in women . From 70–80% of cases of urticarial vasculitis are normocomplementemic and they follow a benign course with an average duration of 3 years.
Pathogenesis
The pathogenesis of urticarial vasculitis is similar to that of typical CSVV. In urticarial vasculitis, it is thought that complement activation triggers mast cell release of inflammatory mediators such as TNF-α that in turn increase the expression of ICAM on mast cells (important for eosinophil transmigration) and E-selectin on endothelial cells .
Although urticarial vasculitis is most often idiopathic, it can be associated with autoimmune connective tissue diseases (especially Sjögren syndrome and SLE), serum sickness, cryoglobulinemia, infections, medications, and hematologic malignancies (see Table 24.5 ).
Clinical features
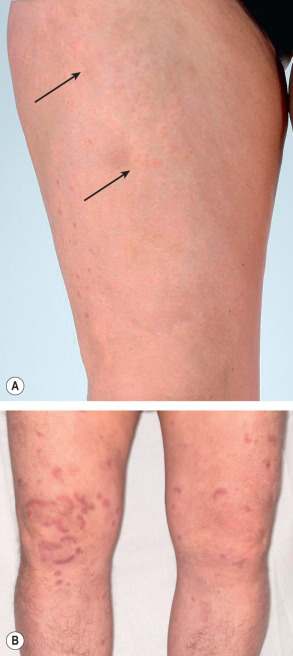
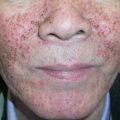
Lesions of urticarial vasculitis consist of erythematous, indurated wheals ( Fig. 24.10 ), with or without angioedema, that favor the trunk and proximal extremities. Urticarial vasculitis is distinguished from chronic urticaria by individual lesions that persist beyond 24 hours, are associated with burning and pain rather than pruritus, and resolve with postinflammatory hyperpigmentation. With diascopy or as residua, hemorrhage can be observed. However, these features are not always present. Rarely, bullae, erythema multiforme-like lesions, livedo reticularis, Raynaud phenomenon, and laryngeal edema are clinical manifestations of urticarial vasculitis .
When faced with urticarial lesions that histopathologically demonstrate features of LCV, the most important prognostic feature is the presence or absence of hypocomplementemia. Patients with normal complement levels tend to have skin-limited disease, whereas those with hypocomplementemia are much more likely to have systemic manifestations . The hypocomplementemic urticarial vasculitis syndrome (HUVS) is a more severe syndrome defined by specific diagnostic criteria . They are as follows:
two major criteria : (1) urticaria for 6 months and (2) hypocomplementemia – plus –
two or more minor criteria : (1) vasculitis on skin biopsy; (2) arthralgia or arthritis; (3) uveitis or episcleritis; (4) glomerulonephritis; (5) recurrent abdominal pain; or (6) positive C1q precipitin test with a low C1q level.
Patients with hypocomplementemia who do not meet the criteria for HUVS are considered to have hypocomplementemic urticarial vasculitis (but not HUVS).
Musculoskeletal involvement is the most common extracutaneous manifestation of urticarial vasculitis. Arthralgias of the hands, elbows, knees, ankles, and feet occur in half of all patients with urticarial vasculitis, but up to 50% of patients with HUVS have frank arthritis . Up to 20% of patients with HUVS have pulmonary symptoms (cough, laryngeal edema, hemoptysis, dyspnea, asthma, chronic obstructive pulmonary disease [COPD]) . COPD is especially severe in smokers with urticarial vasculitis, and it is worse than would be expected due to smoking alone. Renal involvement, manifesting as proteinuria or microscopic hematuria, occurs in 5–10% of patients with HUVS . Gastrointestinal manifestations (abdominal pain, nausea, vomiting, diarrhea) occur in up to 30% of patients; cardiac and central nervous system involvement are rare, but reported. HUVS shares features with SLE, but distinctive clinical findings in HUVS include ocular inflammation (30%; conjunctivitis, episcleritis, iritis, uveitis), angioedema (>50%), and COPD-like symptoms (50%) . Similarly, a recent study of 57 patients with hypocomplementemic urticarial vasculitis found various systemic manifestations including ocular (56%), pulmonary (19%), gastrointestinal (18%), and renal (14%) .
In patients with urticarial vasculitis, the most common abnormal laboratory studies are an elevated ESR, low serum C3 and C4 levels, and a positive ANA. HUVS is marked by low serum complement levels (which may vary, however, from non-detectable to normal, even during attacks) plus the presence of anti-C1q precipitin and depressed C1q levels. Although up to a third of patients with SLE have circulating anti-C1q antibodies and up to half of patients with HUVS have a positive ANA, patients with HUVS rarely have anti-dsDNA or anti-Sm antibodies .
Pathology
Histopathologically, urticarial vasculitis is defined as a minimum of leukocytoclasia with vessel wall necrosis, with or without fibrinoid deposits, perivascular inflammation, or red blood cell extravasation . In one series, an interstitial neutrophilic infiltrate was more commonly observed in patients with the hypocomplementemic form of urticarial vasculitis . Eosinophils may also be noted. Although findings can be subtle and consist only of interstitial neutrophils or perivascular lymphocytes with extravasated erythrocytes (especially in older lesions), these findings alone do not fulfill the histologic criteria for the diagnosis of urticarial vasculitis.
By DIF, 70% of lesions demonstrate deposits of immunoglobulin, C3, or fibrinogen around blood vessels . A granular pattern of immunoreactants along the basement membrane zone occurs in ~80% of lesions and, when accompanied by hypocomplementemia, suggests the diagnosis of SLE . Basement membrane immunoreactants have also been associated with renal disease .
Differential diagnosis
The main entity in the differential diagnosis is urticaria (including delayed pressure urticaria, in which lesions can last longer than 24 hours). Urticarial vasculitis should also be distinguished from neutrophilic urticaria, which, although considered by some to be within the spectrum of urticarial vasculitis, is not associated with hypocomplementemia or autoimmune disease and is better thought of as a subset of urticaria that histologically demonstrates a neutrophilic infiltrate without vasculitis. The differential diagnosis also includes disorders that clinicopathologically present with urticarial lesions and an interstitial neutrophilic infiltrate (i.e. neutrophilic urticarial dermatosis) , including Schnitzler syndrome, adult-onset Still disease, and cryopyrin-associated periodic syndromes (see Ch. 45 ). Additional entities to consider are the urticarial phase of bullous pemphigoid, atypical erythema multiforme, urticaria multiforme , Sweet syndrome, tumid lupus erythematosus, SLE (see above), and rheumatoid neutrophilic dermatitis. For lesions of angioedema, the differential diagnosis includes acquired and inherited forms of angioedema (see Ch. 18 ).
Treatment
No randomized clinical trials have been performed to date to evaluate possible therapeutic options. Antihistamines may reduce the swelling and pain associated with cutaneous lesions, but do not alter the course of the disease. Oral corticosteroids are effective, but the duration of use should be kept to a minimum (see Ch. 125 ). Indomethacin, dapsone (with or without pentoxifylline), colchicine, hydroxychloroquine, and mycophenolate mofetil have all been reported to be beneficial (see Table 24.10 ). Rituximab or intravenous immunoglobulin may also be a useful therapy for recalcitrant hypocomplementemic urticarial vasculitis.
Erythema Elevatum Diutinum
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


