Abstract
Darier disease and Hailey–Hailey disease are autosomal dominant genodermatoses caused by mutations in genes encoding Ca 2+ ATPase pumps in the endoplasmic reticulum (SERCA2) and Golgi apparatus (hSPCA1), respectively. As a consequence, abnormal intracellular Ca 2+ signaling leads to impaired processing of junctional proteins, resulting in acantholysis due to disrupted epidermal adhesion in both disorders and dyskeratosis due to increased apoptosis in Darier disease. Onset of clinical manifestations is often in the second decade of life or later. Darier disease presents with keratotic papules in a seborrheic and acral distribution, whereas Hailey–Hailey disease features blistering and erosions in intertriginous sites. Both disorders are frequently complicated by secondary infections that produce malodor and promote vegetating plaques. Treatment is primarily directed against hyperkeratosis, inflammation, infection and sweating.
Keywords
Darier disease, Hailey–Hailey disease, acrokeratosis verruciformis of Hopf, acantholysis, dyskeratosis
Darier Disease
▪ Darier–White disease ▪ Keratosis follicularis ▪ Dyskeratosis follicularis
- ▪
Autosomal dominant disorder in which mutations in the ATP2A2 gene result in dysfunction of an endoplasmic reticulum Ca 2+ ATPase (SERCA2), thus interfering with intracellular Ca 2+ signaling
- ▪
Keratotic and crusted papules and plaques favor seborrheic areas; additional features include palmoplantar papules, variable nail changes, and whitish oral mucosal papules
- ▪
Clinical subtypes include an acral hemorrhagic variant and segmental forms due to type 1 and type 2 mosaicism
- ▪
Characterized histologically by acantholytic dyskeratosis with suprabasilar clefting, “corps ronds” and “grains”
Introduction
Darier disease is an autosomal dominant genodermatosis with characteristic mucocutaneous findings such as keratotic papules on the upper trunk and longitudinal erythronychia. Insufficient function of the 2b isoform of the sarco/endoplasmic reticulum Ca 2+ ATPase (SERCA2b) leads to abnormal intracellular Ca 2+ signaling, notably involving the endoplasmic reticulum. The result is a loss of suprabasilar cell adhesion (acantholysis) and an induction of apoptosis (dyskeratosis).
History
In 1889, the French dermatologist Jean Darier , at the Hôpital Saint-Louis in Paris, and James C White , Professor of Dermatology at Harvard University, independently reported a skin disease characterized by brown, crusted, malodorous lesions with a follicular distribution. Darier called the disease “psorospermose folliculaire végétante” because of his histopathologic studies, where throughout the malpighian layer he saw a large number of round bodies surrounded by a double-layered membrane. He and others thought that these bodies represented psorosperms, a type of parasitic protozoa now referred to as coccidia, which they postulated were the causative agents for the disease. However, attempts to culture or inoculate the supposed parasites failed, and the histologic changes were subsequently recognized as sequelae of abnormal keratinization . The supposed follicular origin of the disease could not be confirmed either, as lesions were noted to occur outside the pilosebaceous unit . It was White who first suggested the hereditary character of the disorder, when the daughter of his initially described patient developed similar skin lesions .
Epidemiology
The prevalence of Darier disease has ranged from 1 in 100 000 in Denmark to 1 in 30 000 in Scotland, and the estimated incidence is 4 per million per 10 years . Men and women are equally affected. Darier disease has an autosomal dominant mode of inheritance with complete penetrance and variable expressivity. For example, in a series of 42 patients with Darier disease, 7 of 18 affected individuals who recalled no family history were found to be members of known Darier kindreds with mildly affected parents . The clinical severity varies among different families carrying identical mutations and among members of the same family. Indeed, subtle changes of the skin or nails may remain unnoticed by affected persons.
Pathogenesis
Mutations in the ATP2A2 gene which encodes an endoplasmic reticulum (ER) Ca 2+ ATPase pump , SERCA2, leads to both acantholysis and apoptosis, accounting for the characteristic histologic finding of acantholytic dyskeratosis. SERCA2 haploinsufficiency has been hypothesized to underlie the condition’s dominant inheritance pattern , but a more recent study found that aggregates of mutant SERCA2 may exert a dominant negative effect by inducing ER stress and keratinocyte apoptosis . Although both SERCA2a and SERCA2b isoforms are found within the epidermis, SERCA2b is the major protein product and its dysfunction alone can produce Darier disease . While SERCA2 is ubiquitously expressed, a lack of compensatory SERCA3 expression in keratinocytes may explain the particular vulnerability of these cells to SERCA2 deficiency.
More than 240 pathogenic ATP2A2 mutations have been identified, primarily missense and frameshift alterations. In general, there is no clear genotype–phenotype correlation . Possible exceptions include particular ATP2A2 mutations associated with acrokeratosis verruciformis of Hopf and the acral hemorrhagic subtype of Darier disease . Several pathogenic mechanisms have been proposed for the acantholytic dyskeratosis of Darier disease. ATP2A2 mutations result in chronically low Ca 2+ stores within the ER ( Fig. 59.1 ) , which is aggravated by cellular stress. This Ca 2+ depletion leads to acantholysis via impaired processing, folding and trafficking of junctional proteins such as desmoplakin and E-cadherin . Impaired membrane translocation of protein kinase C α, an important Ca 2+ -dependent regulatory enzyme that interacts with membrane phospholipids, is thought to mediate the disruption of desmosome assembly . Recent studies have also shown that SERCA2 inhibition in keratinocytes causes increased levels of sphingosine, which inhibits protein kinase C α .
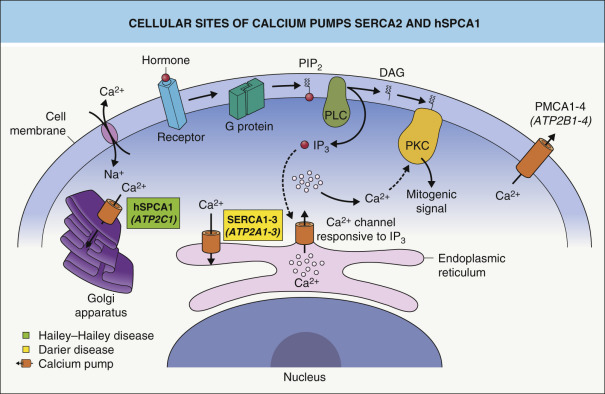
ER stress due to depleted Ca 2+ stores and accumulation of unfolded proteins produces the “unfolded protein response”, which can induce apoptosis . ATP receptors, which are thought to have roles in apoptosis and calcium signaling, were reported to localize abnormally in lesional Darier disease epidermis, with decreased P2Y2 (a G protein-coupled receptor) but increased P2X7 (a death receptor) in the plasma membrane of acantholytic cells .
Clinical Features
Onset and clinical pattern
In approximately 70% of patients, the disease begins between the ages of 6 and 20 years, with a peak onset during puberty (ages 11–15 years) . The primary lesions are keratotic, sometimes crusted, red to brown papules, which develop preferentially in a “seborrheic” distribution involving the trunk ( Fig. 59.2 ), scalp (especially its margins; Fig. 59.3 ), face, and lateral aspects of the neck ( Fig. 59.4 ). Despite its initial description as follicular dyskeratosis, the papules are not limited to a perifollicular location. The lesions tend to become confluent and may form papillomatous masses ( Fig. 59.2B,D ). Small (2–3 mm) hypomelanotic macules may be admixed with the keratotic papules and occasionally are the predominant feature ( Fig. 59.5 ). Rarely, sterile vesicles or bullae are prominent, and, in these cases, herpetic infection (Kaposi varicelliform eruption) must be excluded .
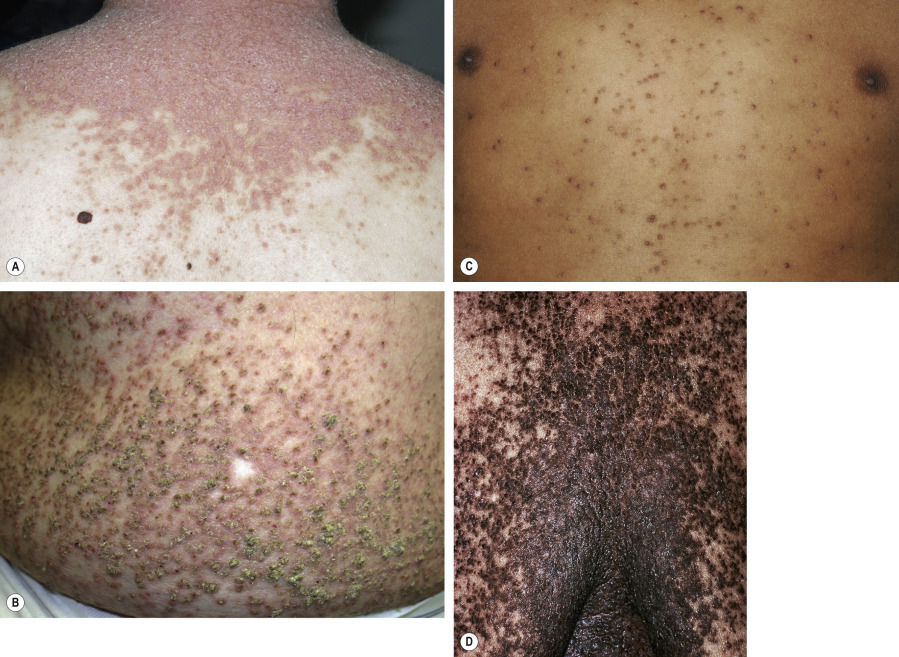
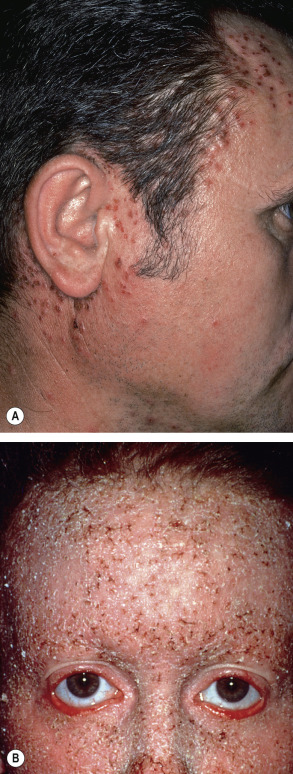
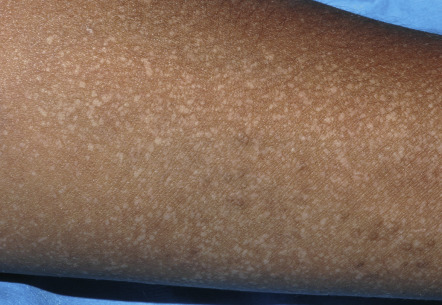
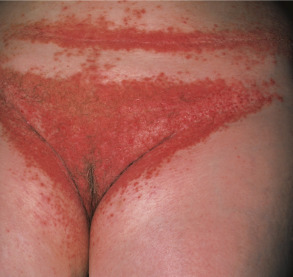
Most patients have lesions in the axillae, groin, and/or inframammary area ( Fig. 59.6 ). Although these intertriginous lesions are usually mild, they are occasionally the predominant finding and may result in the misdiagnosis of Hailey–Hailey disease (HHD) . Occasionally, macerated, fungating masses are seen in the axillae or groin. Malodor is a frequent and distressing feature of Darier disease.
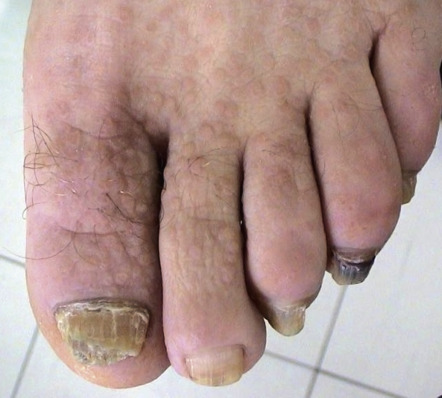
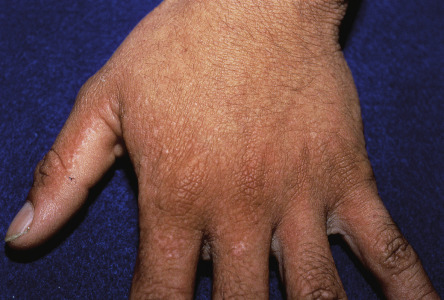
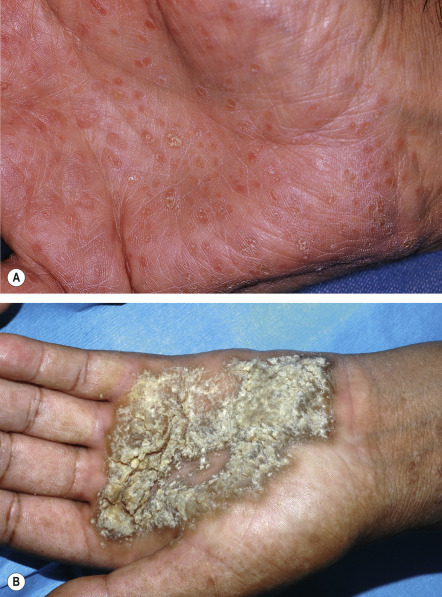
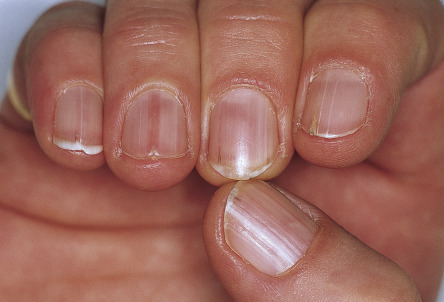
In ~50% of patients, 2–4 mm, skin-colored or brownish, flat-topped papules, reminiscent of flat warts, are found on the dorsal aspects of the hands and feet, and less often, on the forearms and legs ( Fig. 59.7 ). Rarely, acral hemorrhagic vesicles may also be seen. Palmoplantar papules, many of which are keratotic, and keratin-filled depressions are almost invariably present ( Fig. 59.8 ). Subtle changes can be detected by obtaining finger or palm prints, which show interruption of the dermatoglyphic pattern. Nail changes include longitudinal red and/or white lines, longitudinal ridging and fissuring, and wedge-shaped subungual hyperkeratosis ( Fig. 59.9 ). The nails are brittle and tend to break distally, forming V-shaped notches .
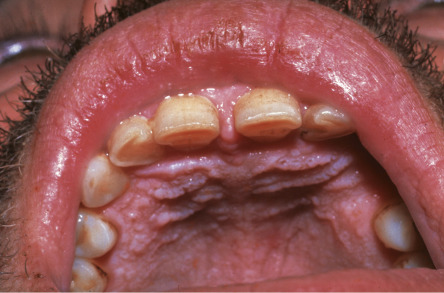
Painless whitish papules or rugose plaques are noted in 15–50% of patients with Darier disease. The hard palate is the most common site of involvement, followed by the gingiva, buccal mucosa, and tongue ( Fig. 59.10 ) .
Symptoms
Most patients complain of moderate itching. Equally distressing are the appearance and the odor, which can lead to social isolation.
Modifying factors
This condition frequently worsens during the summer. In patients with Darier disease, characteristic lesions can be experimentally induced in clinically uninvolved skin by UV irradiation, and UVB irradiation has been shown to suppress ATP2A2 expression in keratinocytes . Sweating, heat, and occlusion represent additional factors that trigger exacerbations primarily in covered and intertriginous areas (e.g. after a long airplane flight), and this predisposes patients to infections (see below). Lithium carbonate, which is commonly used to treat bipolar disorder , can provoke flares of Darier disease.
Course of the disease
Darier disease follows a chronic course with fluctuations in disease severity. While some patients report improvement over time, others experience a worsening .
Infectious complications
Areas of skin affected by Darier disease are prone to secondary infections with bacteria, yeast, and dermatophytes , often leading to malodor and vegetating plaques. A study of 75 adult patients found a high prevalence of Staphylococcus aureus colonization in lesional skin (~70%) and the nares (~50%), which correlated with a more severe phenotype .
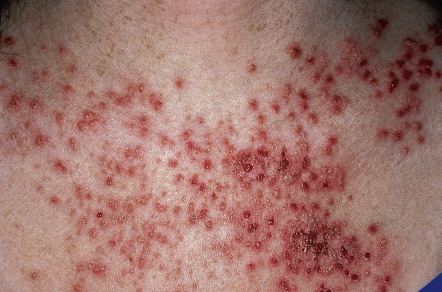
Affected individuals are also susceptible to the development of widespread cutaneous infections with human papillomaviruses (HPV) and herpes simplex virus (HSV). When there is a sudden onset and rapid spread of vesicular and crusted lesions, often accompanied by fever and malaise, the clinician should have a high index of suspicion for the development of Kaposi varicelliform eruption ( Fig. 59.11 ) . The latter requires systemic antiviral therapy (e.g. acyclovir, valacyclovir). Generalized cowpox infection has also been reported.
Disruption of the epidermal barrier likely plays an important role in the increased frequency and severity of skin infections in patients with Darier disease. Immunologic studies have shown no consistent systemic abnormalities . However, a immunohistochemical analysis found evidence for an impaired local immune response, with a decrease in Langerhans cells and plasmacytoid dendritic cells .
Salivary glands
In some patients, obstruction of salivary gland ducts can lead to painful glandular swelling. Darier-like histologic changes within the ducts are thought to be responsible for the obstruction .
Neuropsychiatric disorders
Various neuropsychiatric conditions such as epilepsy, intellectual impairment, and mood disorders have been reported in association with Darier disease, but the strength and nature of these relationships remain to be determined . Neuropsychiatric assessment of 100 Darier disease patients from the UK revealed higher lifetime rates of major depression (30%), suicide attempts (13%), bipolar disorder (4%), and epilepsy (3%) than in the general population . In a recent study based on the Swedish national registry, Darier disease patients (n=770) were more likely to be diagnosed with intellectual disability (6.2-fold), bipolar disorder (4.3-fold), and schizophrenia (2.3-fold) than matched individuals from the general population .
There are no known direct neuropsychiatric effects of ATP2A2 mutations , although an accumulation of insoluble SERCA2 aggregates within neurons has been hypothesized . It is also possible that there may be a closely linked susceptibility gene for bipolar disorder . In addition, the disfigurement and isolation associated with severe skin diseases often contribute to psychiatric and social morbidity. Therefore, depression and increased suicidal ideation in patients with Darier disease may be explained, at least in part, by non-genetic factors .
Others
Ocular complications, e.g. corneal ulcerations or staphylococcal endophthalmitis, occur very rarely. There have been a few reports of squamous cell carcinomas arising in cutaneous or mucosal sites chronically affected by Darier disease . Malignant transformation is a rare event and may be related to infection with oncogenic types of HPV or changes in keratinocyte adhesion and proliferation secondary to SERCA2 haploinsufficiency, which has been associated with squamous cell carcinomas of the skin and upper gastrointestinal tract in mice. Observations of bone cysts, renal agenesis, and autoimmune thyroiditis in patients with Darier disease are thought to represent chance associations.
Clinical subtypes of Darier disease ( Table 59.1 )
Acral hemorrhagic type
In addition to classic clinical features, a small subset of patients develop sharply demarcated, red to blue–black macules on the palms and soles as well as the dorsal aspects of the hands . These irregularly shaped lesions represent hemorrhage into acantholytic vesicles. A particular ATP2A2 missense mutation (N767S) has been identified in several non-related families with the acral hemorrhagic type of Darier disease , as well as a few individuals with a classic Darier disease phenotype .
| LESS COMMON PRESENTATIONS OF DARIER DISEASE |
| Morphologic variants |
|
| Distribution patterns |
|
| Extracutaneous |
|
* A persistent acantholytic dermatosis and extensive lentiginous “freckling” without an underlying ATP2A2 mutation has also been described.
† The differential diagnosis may include familial dyskeratotic comedones.
Segmental types 1 and 2
Two types of segmental Darier disease have been described, both with a distribution of lesions along the lines of Blaschko (see Ch. 62 ). With the more common type 1 mosaicism ( Fig. 59.12 ), the age of onset, severity, and histologic findings within the streaks are similar to those of generalized Darier disease. Type 1 segmental Darier disease results from a postzygotic mutation in the ATP2A2 gene during embryogenesis, which leads to a mosaic pattern of skin involvement. Heterozygous ATP2A2 mutations in affected, but not background, skin have been documented in patients with this form of segmental Darier disease . If there are mutant cells in the gonads, a patient with type 1 segmental manifestations may have offspring with generalized Darier disease.
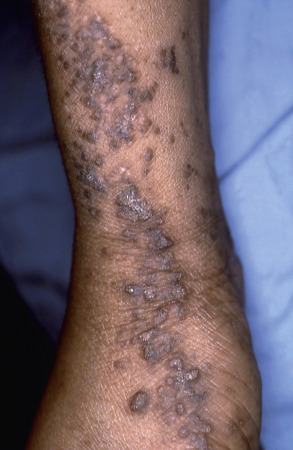
Patients with type 2 mosaicism have generalized Darier disease plus linear streak(s) with increased severity . Type 2 segmental manifestations of autosomal dominant disorders occur when patients with a heterozygous germline mutation also have a postzygotic inactivating mutation in the other allele of the same gene (i.e. a “second hit” causing loss of heterozygosity), which leads to more severe manifestations in a mosaic distribution (see Fig. 62.2 ). Of note, a second hit in the ATP2A2 gene was identified in skin from erosive patches following the lines of Blaschko in a 1-year-old boy with a family history of Darier disease .
Pathology
There are two prominent histologic features in Darier disease, acantholysis and dyskeratosis ( Fig. 59.13 ). Acantholysis is due to a disturbance in cell adhesion that leads to suprabasilar cleft formation. At the ultrastructural level, this corresponds to a loss of desmosomes and detachment of keratin filaments from the desmosomes . Dyskeratosis is due to apoptosis of keratinocytes and is characterized by nuclear condensation and perinuclear keratin clumping. Two types of dyskeratotic cells are observed in Darier disease :
- •
“Corps ronds” – acantholytic enlarged keratinocytes in the malpighian layer with darkly staining and partially fragmented nuclei surrounded by a clear cytoplasm and encircled by a bright ring of collapsed keratin bundles.
- •
“Grains” – small, oval cells in the stratum corneum characterized by an intensely eosinophilic cytoplasm composed of collapsed keratin bundles containing shrunken parakeratotic nuclear remnants. “Grains” are likely derived from “corps ronds”, but formal proof that they indeed represent different stages of the same pathologic process has not been provided.
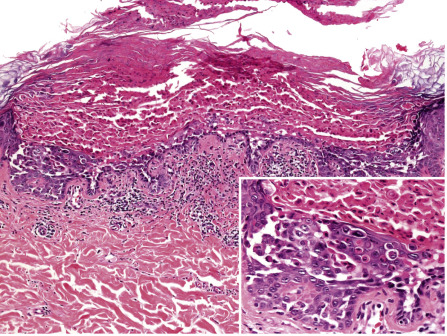
The epidermis overlying acantholytic and dyskeratotic foci is thickened and shows papillomatosis and hyperkeratosis. In the superficial dermis, there is a mild to moderate perivascular inflammatory infiltrate. Diagnostic histologic changes are often focal, necessitating a careful search. Similar histologic features may be observed in Grover disease, but in the latter there tends to be more acantholysis, less dyskeratosis, and fewer “corps ronds” or “grains”. The two conditions can be histologically indistinguishable, but the findings are typically more pronounced, widespread, and follicle-based in Darier disease.
Differential Diagnosis
Acrokeratosis verruciformis of Hopf
Flat-topped, wart-like papules on the dorsal aspects of the extremities are a common manifestation of Darier disease. They are clinically indistinguishable from the lesions that characterize the autosomal dominant disorder described by Hopf as “acrokeratosis verruciformis” ( Fig. 59.14 ) . There has been debate as to whether acrokeratosis verruciformis is a separate entity or an allelic forme fruste of Darier disease, and reports of an underlying ATP2A2 mutation in some, but not all, patients with acrokeratosis verruciformis suggest genetic heterogeneity . Acrokeratosis verruciformis and Darier disease have traditionally been separated on the basis of their histologic features, with the former lacking acantholysis and dyskeratosis. However, serial sectioning is often required to find foci of acantholysis and dyskeratosis in acral skin biopsy specimens from patients with Darier disease .