Abstract
This chapter covers the histiocytoses with emphasis on Langerhans cell histiocytosis (LCH) and many of the disorders comprising the large group of diseases referred to as non-Langerhans cell histiocytoses (non-LCH). Both groups represent a spectrum of disease, from self-resolving and skin limited to severe multi-organ involvement with significant morbidity. The immunophenotype of pathologic cells is the key feature in discriminating between LCH and non-LCH. In brief, LCH cells are always S100-, CD1a-, and Langerin (CD207)-positive and usually CD68-negative. The cells in non-LCH disorders are always positive for CD68, always negative for Langerin, and usually negative for S100 and CD1a (although there are exceptions). The major features of the rare malignant histiocytic disorders and even rarer dendritic cell hamartomas are also reviewed.
Keywords
Langerhans cell histiocytosis (LCH), non-Langerhans cell histiocytosis (non-LCH), CD1a, CD68, Langerin (CD207), BRAF V600E mutation, congenital self-healing reticulohistiocytosis (Hashimoto–Pritzker disease), juvenile xanthogranuloma, necrobiotic xanthogranuloma, reticulohistiocytosis, Rosai–Dorfman disease, xanthoma disseminatum, malignant histiocytic disorders, dermal dendritic hamartoma
Overview
The histiocytoses represent a group of proliferative disorders that share a common CD34 + progenitor cell in the bone marrow. Three “histiocytes” of cutaneous importance are: (1) the Langerhans cell , which migrates to and from the epidermis and functions as a potent antigen-presenting cell (APC); (2) the mononuclear cell/macrophage (sometimes called the “true” histiocyte), which migrates to and from the dermis and has both phagocytic and APC properties; and (3) the dermal dendrocyte/dendritic cell , of which there are two subtypes. Type 1 dermal dendrocytes are factor XIIIa-positive and generally reside within the papillary dermis, whereas type 2 dermal dendrocytes are CD34-positive and are generally found within the reticular dermis. Type 1 dermal dendrocytes may be involved in phagocytosis, antigen presentation, inflammation, collagen production and wound healing, while the function of type 2 dermal dendrocytes is less certain. These two types represent myeloid dendritic cells and are distinct from plasmacytoid dendritic cells.
Dysfunction of these various histiocytes has led to a group of well-known, but poorly understood, disorders. For years, many of the histiocytoses were known by numerous names, reflecting the lack of understanding and agreement regarding their origin. Electron microscopy and the development of immunohistochemical stains have since provided insight into these conditions. It is now clear that the histiocytoses are closely related entities, with Langerhans cell histiocytosis (LCH) representing one group of disorders and non-Langerhans cell histiocytosis (non-LCH) representing another. Of note, each group represents a spectrum of disease, with overlap between entities.
This chapter covers the most common and several of the rarer histiocytic disorders in detail ( Table 91.1 ). In addition, the rare malignant histiocytic disorders and the even rarer dendritic cell hamartomas are reviewed.
| CLINICAL FEATURES OF THE HISTIOCYTOSES |  | |||
|---|---|---|---|---|
| Histiocytosis | Usual age | Most common mucocutaneous sites | Other findings | |
| Langerhans cell histiocytoses | ||||
| Letterer–Siwe disease | 0–2 years | Scalp, flexural areas, trunk | Visceral and bone lesions | |
| Hand–Schüller–Christian disease | 2–6 years | Scalp, flexural areas, trunk, gingival | Diabetes insipidus, bone lesions, exophthalmos | |
| Eosinophilic granuloma | 7–12 years | Skin lesions rare | Bone lesions primarily | |
| Congenital self-healing reticulohistiocytosis (Hashimoto–Pritzker disease) | Congenital | Widespread, localized, or single lesion | Spontaneous resolution, but children should be followed longitudinally | |
| Non-Langerhans cell histiocytoses | ||||
| Primarily cutaneous, usually self-resolving | ||||
| Juvenile xanthogranuloma | 0–2 years | One to a few lesions ≫ numerous & widespread Head and neck > upper trunk > extremities | Rare eye and visceral lesions; spontaneous resolution; association with CALM, NF1, and/or juvenile myelomonocytic leukemia | |
| Benign cephalic histiocytosis | 0–3 years | Face and neck > trunk and extremities | Usually none (diabetes insipidus rare); spontaneous resolution | |
| Giant cell reticulohistiocytoma | Adults | Head (solitary lesion) | None; spontaneous resolution | |
| Generalized eruptive histiocytoma | <4 and 20–50 years | Widespread (axial) | Recurrent crops; spontaneous resolution | |
| Indeterminate cell histiocytosis | Any | Solitary or generalized Trunk & extremities > head & neck, genitalia | Uncommon visceral and bone lesions; possible association with B-cell lymphoma and leukemia | |
| Primarily cutaneous, often persistent/progressive | ||||
| Papular xanthoma * | Any | Generalized (discrete yellow papules and papulonodules with relative sparing of flexural sites) | Mucous membrane involvement can occur occasionally | |
| Progressive nodular histiocytoma * | Any | Generalized (discrete yellow papules and nodules, sometimes with prominent facial involvement) | May represent same entity as the progressive form of papular xanthoma ; mucous membrane involvement can occur | |
| Hereditary progressive mucinous histiocytosis * | Childhood/adolescence | Generalized (skin-colored to erythematous papules and nodules) | Usually occurs in female patients; histologically, abundant dermal mucin in addition to histiocytes | |
| Cutaneous, with frequent systemic involvement | ||||
| Necrobiotic xanthogranuloma | 17–60 years | Periorbital > other face, trunk, extremities | Paraproteinemia due to plasma cell dyscrasia or lymphoproliferative disorder, hepatosplenomegaly | |
| Multicentric reticulohistiocytosis | 30–50 years | Head; hands, elbows (over joints); mucosa (oral, nasopharyngeal) | Arthritis (often destructive); up to 30% with internal malignancy | |
| Rosai–Dorfman disease | 10–30 years | Eyelids and malar area | Massive lymphadenopathy in a subset of patients, fever, hypergammaglobulinemia; skin-limited form increasingly recognized | |
| Xanthoma disseminatum | Any | Flexural areas to widespread > mucosa (oral, nasopharyngeal) | Diabetes insipidus | |
| Systemic, with skin involvement rare to unusual | ||||
| Erdheim–Chester disease * | Any, but usually adults | Skin involvement ~25% of patients: eyelids, scalp, neck, trunk, axillae (red–brown to yellow nodules and indurated plaques) | Fever, bone pain (osteosclerosis of long bones), exophthalmos, diabetes insipidus; involvement of the lungs, kidneys, adrenals, heart (up to 50%), CNS, retroperitoneum (fibrosis), and testes; high mortality rate; ~55% of patients have BRAF V600E mutations Foamy histiocytes with a small nucleus (CD68 + . CD163 + , CD1a − ); a few multinucleated histiocytes or Touton giant cells also often present | |
Sea-blue histiocyte syndrome (sea-blue histiocytosis) *
| Usually adolescence/young adulthood (inherited form) | Skin involvement rare (inherited form): facial (macular hyperpigmentation and nodules) | Histiocytes contain cytoplasmic granules that stain azure blue with May–Gruenwald stain; multiple organs involved; can be fatal | |
Hemophagocytic lymphohistiocytosis (HLH; hemophagocytic syndrome) *
| Primary: 0–2 years Secondary: any | Generalized (purpuric macules and papules, morbilliform eruptions, erythroderma, keratotic nodules) or acral (erythematous macules); malignancy-associated forms may present with lesions of cutaneous/subcutaneous cytotoxic NK/T-cell lymphomas; skin biopsy specimens rarely demonstrate hemophagocytosis |
| |
* Not covered in the body of the text.
‡ In the US, most commonly due to mutations in the gene that encodes perforin ( PRF1 ), referred to as subtype 2; additional genes include UNC13D (subtype 3), STX11 (subtype 4), and STXBP2 (subtype 5). Their protein products play a role in the cytotoxic activity of lymphocytes, including the exocytosis of cytotoxic granules by NK cells.
** The primary differential diagnosis is iron overload, Still disease, HLH or MAS, infection, and malignancy.
Langerhans Cell Histiocytosis
▪Langerhans cell histiocytosis (LCH; class I histiocytosis, histiocytosis X), comprising:
- ▪
Letterer–Siwe disease
- ▪
Hand–Schüller–Christian disease
- ▪
Eosinophilic granuloma
- ▪
Congenital self-healing reticulohistiocytosis (Hashimoto–Pritzker disease)
- ▪
LCH is a clonal proliferative disorder
- ▪
By immunohistochemistry, LCH cells are positive for S100 protein, CD1a and Langerin (CD207)
- ▪
Ultrastructurally, intracytoplasmic Birbeck granules are seen
- ▪
Systemic manifestations include osteolytic bone lesions and diabetes insipidus
Introduction
Langerhans cell histiocytosis (LCH) is a clonal proliferative disorder of Langerhans cells that express an immunophenotype positive for S100 protein, CD1a, and Langerin (CD207) and contain cytoplasmic Birbeck granules. LCH represents a disease spectrum with four historically distinct but actually overlapping syndromes:
- •
Letterer–Siwe disease
- •
Hand–Schüller–Christian disease
- •
eosinophilic granuloma
- •
congenital self-healing reticulohistiocytosis (Hashimoto–Pritzker disease).
Disease expression ranges from mild, sometimes asymptomatic, single-organ involvement to severe, progressive multisystem disease.
History
Letterer–Siwe disease, Hand–Schüller–Christian disease, and eosinophilic granuloma were described in the early twentieth century. In 1953, Lichtenstein grouped the three disorders into a single entity which he called histiocytosis X . Twenty years later, Hashimoto and Pritzker described the entity congenital self-healing reticulohistiocytosis . Immunologic and ultrastructural studies confirmed the relationship of the pathologic cells in histiocytosis X and congenital self-healing reticulohistiocytosis to Langerhans cells, providing a basis for the Writing Group of the Histiocyte Society, in 1987, to reclassify them as “Langerhans cell histiocytoses” .
Epidemiology
LCH occurs worldwide and most commonly develops in children ages 1–3 years, although disease can develop at any age. The reported incidence of LCH varies widely. However, an annual incidence of at least five per million children is often quoted, with the adult incidence suspected to be less than one-third that of children. LCH is more common in boys, with a male : female ratio of nearly 2 : 1. In adults, there may be a slight female predominance.
Some cases of LCH appear to be familial. One study described simultaneous development of LCH in four of five monozygotic twin pairs and in one of three dizygotic pairs . Additionally, in two families with affected non-twin siblings, parental consanguinity was known in one and possible in the other.
Pathogenesis
Until recently, the pathogenesis of LCH was unknown. Although viral and immunologic etiologies had long been considered, it is more likely that the latter, e.g. elevated lesional cytokine levels, play a secondary role. In 2010, a significant percentage (55–60%) of LCH specimens were shown to harbor the BRAF V600E mutation , as has been described in cutaneous melanomas and papillary thyroid carcinomas. This finding strongly suggested that LCH is neoplastic and raised the possibility of the therapeutic use of BRAF inhibitors.
As mentioned previously, at least a small subset of LCH appears to be hereditary . In addition, several studies by different investigators have demonstrated clonal CD1a + histiocytes in all LCH tissue tested . Based upon these results, as well as the presence of oncogenic BRAF mutations, LCH is now best regarded as a clonal neoplastic disorder. Whether the cell of origin is an immature myeloid dendritic cell or a mature tissue-based dendritic cell remains to be determined.
Clinical features
The four well-described variants of LCH have significant clinical overlap. As a result, many clinicians no longer attempt to differentiate between the various syndromes that comprise LCH, recognizing that LCH is one disease with a wide clinical spectrum and a highly varied course. For historical context, the three classically described variants and the more recently recognized congenital self-healing reticulohistiocytosis are discussed here.
Letterer–Siwe disease is the acute diffuse form of LCH. It is a multisystem disease that nearly always develops prior to age 2 years, and commonly presents in children less than 1 year of age. Cutaneous involvement occurs in most patients as 1–2 mm pink to skin-colored papules, pustules and/or vesicles in the scalp, flexural areas of the neck, axilla and perineum, and on the trunk ( Fig. 91.1A,B,C ). Rarely, the lesions resemble mollusca contagiosa ( Fig. 91.1D ) . Scale and crust with secondary impetiginization are common findings, as is the development of petechiae and purpura ( Fig. 91.1E ). The lesions tend to coalesce and become tender, especially when fissures develop in intertriginous zones ( Fig. 91.1B,F ). Palmoplantar and nail involvement can occur, as can soft tissue nodules ( Fig. 91.1G ). In patients with darkly pigmented skin, the lesions may appear relatively hypopigmented ( Fig. 91.1H ). The eruption is most often confused with seborrheic dermatitis, various forms of diaper dermatitis or intertrigo (see Figs 13.3 and 13.11 ), arthropod bites (including scabies), and varicella.
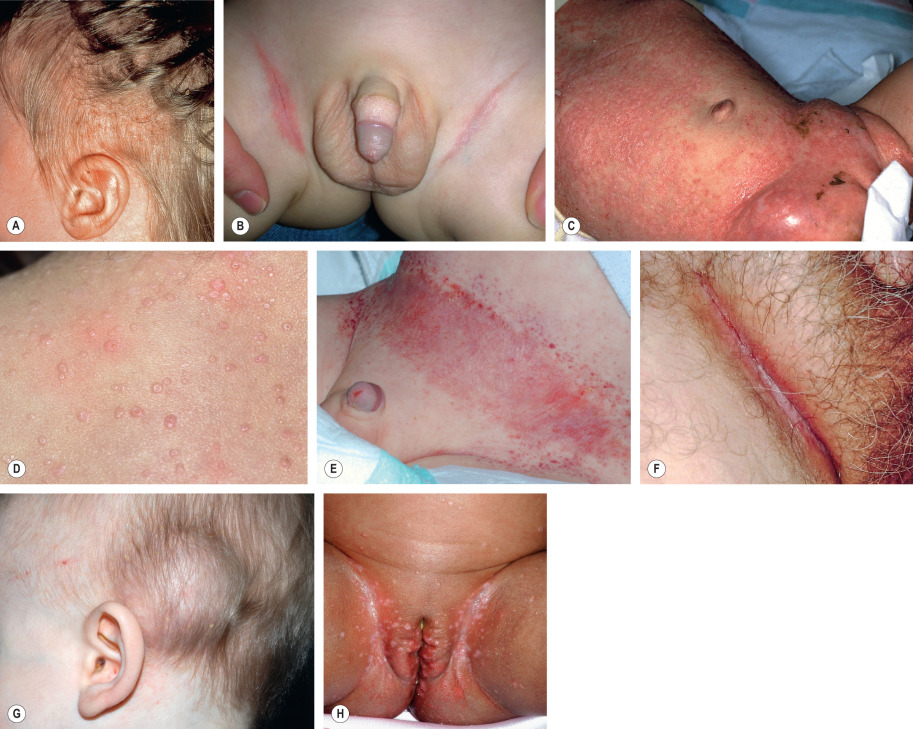
During the course of the disease, many organs can become infiltrated by clonal LCH cells. However, only if the key functions of the organ are affected is such involvement of prognostic significance. Lung, liver, lymph node, and bone involvement commonly occur at some point during the illness. Osteolytic bone lesions are painful, usually multiple, and most frequently involve the cranium. Occasionally, the hematopoietic system can be affected, with thrombocytopenia and anemia portending a poor prognosis.
Classically, Hand–Schüller–Christian disease represents the triad of diabetes insipidus, bone lesions, and exophthalmos. These patients tend to have a chronic, progressive course. Typically, Hand–Schüller–Christian disease begins between the ages of 2 and 6 years. Patients with the complete triad are rare, as exophthalmos is uncommon and often a late finding. Approximately 30% of patients develop skin or mucous membrane lesions. While early cutaneous lesions are similar to those seen in Letterer–Siwe disease, older lesions can become xanthomatous. Ulcerative nodules may develop in the oral and genital areas, with premature loss of teeth possible secondary to gingival lesions.
At least 80% of patients with Hand–Schüller–Christian disease develop bone lesions, the cranium being preferentially involved. Chronic otitis media occurs commonly in these patients and in patients with all forms of LCH. Diabetes insipidus, secondary to infiltration of the posterior pituitary by LCH cells, develops in approximately 30% of patients and is more common in those individuals with cranial bone involvement. The chances of reversing the diabetes insipidus with radiation or chemotherapy are remote once symptoms develop. However, symptomatic treatment with vasopressin is effective.
Eosinophilic granuloma is a localized variant of LCH that generally affects older children, boys more than girls. Skin and mucous membrane lesions are rare, with a single asymptomatic granulomatous lesion of the bone the most common manifestation. The cranium is most frequently affected, though lesions can also develop within the ribs, vertebrae, pelvis, scapulae, and long bones. A spontaneous fracture or otitis media may be the first sign of disease.
Congenital self-healing reticulohistiocytosis (Hashimoto–Pritzker disease) is a variant of LCH which is generally limited to the skin and rapidly self-healing ( Fig. 91.2 ). It presents at birth or in the first few days of life with a characteristic eruption of widespread red to purplish-brown papulonodules, which may have a vascular appearance or resemble a “blueberry muffin” rash (see Ch. 121 ). After several weeks to months, the lesions crust and involute. Solitary papules or nodules (often eroded or ulcerated) and disseminated vesicular eruptions have also been observed. Mucous membrane lesions and systemic involvement occasionally occur, including later-onset diabetes insipidus. While congenital self-healing reticulohistiocytosis is usually a benign, self-resolving disorder, its relationship to other LCH variants suggests a cautious approach with respect to prognosis, and longitudinal evaluation is recommended.
Adults rarely develop LCH, but when they do, the most commonly involved sites are the skin, lung, and bone (see Fig. 91.1F ). Diabetes insipidus can also develop and, as in children, is more likely when bony involvement of the skull is present. Severe multisystem disease, classically referred to as Letterer–Siwe disease, is rare in adults. However, LCH can be a progressive disease in adults, especially when both bone and extraskeletal sites are involved. Pulmonary involvement can be isolated or be a component of multisystem disease and it favors men who smoke cigarettes .
There are two patterns of associations between LCH and malignancy . The first is the development of acute leukemias and solid tumors in patients with a history of LCH who have been treated with chemotherapy, radiotherapy or both, and the malignancies are likely to be treatment-related. There are reports of a few patients who developed skin or solid tumors (e.g. basal cell carcinoma, osteosarcoma) within the fields of radiotherapy for their LCH.
A second, more fascinating association is the apparent increased incidence of LCH in patients with hematologic malignancies, including acute myelogenous leukemia, acute lymphocytic leukemia, chronic myelomonocytic leukemia, and lymphomas of both T- and B-cell origin. These malignancies can occur prior to, concurrently, or following the diagnosis of LCH . In contrast to treatment-related (often referred to as secondary) hematologic malignancies, it has been demonstrated in several patients that the LCH and the hematologic malignancy are clonally related, possibly suggesting origin from a common neoplastic stem cell .
The prognosis of patients with LCH varies dramatically. Involvement of “risk organs” – hematopoietic system, liver, lungs and/or spleen – substantially increases the risk of disease-related mortality. However, in individuals with single-system disease or multisystem disease that does not involve risk organs, mortality rates are very low (e.g. <5% in the latter group). As with hematologic malignancies, the response to initial systemic treatment is an important prognostic indicator in LCH. Of note, in one study patients with high-risk LCH had BRAF V600E mutations in their CD34 + hematopoietic progenitor cells, while the mutation was limited to lesional CD207 + (Langerin + ) dendritic cells in low-risk LCH patients .
Pathology
Just as the clinical appearance of LCH is variable, so is the histologic appearance. In a typical papule, a proliferation of LCH cells is present in the papillary dermis ( Fig. 91.3A ). These cells are large, 10–15 microns in diameter, with a reniform (kidney-shaped) nucleus. Usually, some epidermal infiltration by Langerhans cells is present, and there may even be aggregates within the epidermis ( Fig. 91.3B ), as well as interface changes. The dermal LCH cells are often admixed with eosinophils, neutrophils, lymphocytes, mast cells, and plasma cells. Secondary features such as crusting, pustule formation, hemorrhage or necrosis may obscure the characteristic LCH cell infiltrate. Older lesions that are no longer proliferative may appear granulomatous, xanthomatous or fibrous.
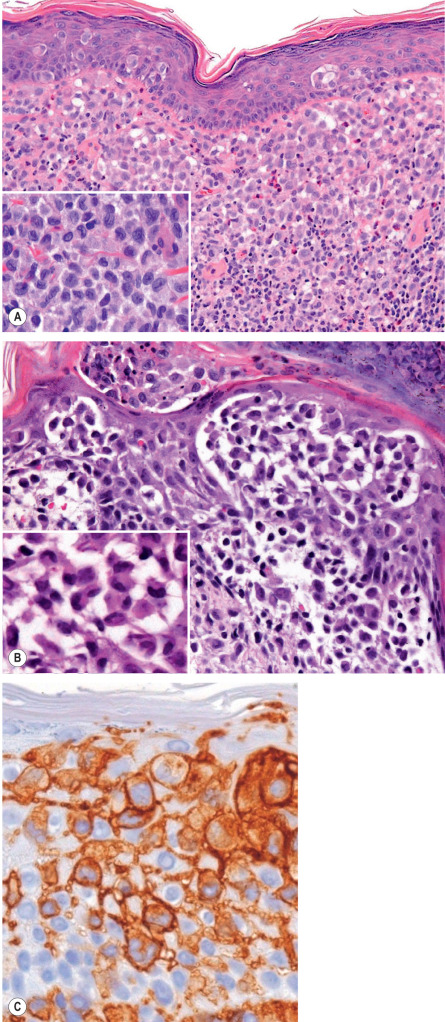
In nodular lesions of congenital self-healing reticulohistiocytosis, the histology may be identical to other variants of LCH. However, lesions with sheets of histiocytes with abundant eosinophilic cytoplasm, so-called “reticulohistiocytes”, intermixed with LCH cells and giant cells, have also been described.
In soft tissue nodules or bone lesions, LCH cells are readily identifiable, especially during the proliferative phase of the process, as the aggregates tend to be larger. In these lesions, sheets of LCH cells may be found, and a few mitoses may be seen. Older bone lesions, on the other hand, may show only xanthomatous or fibrous changes. Early lesions should be biopsied when possible.
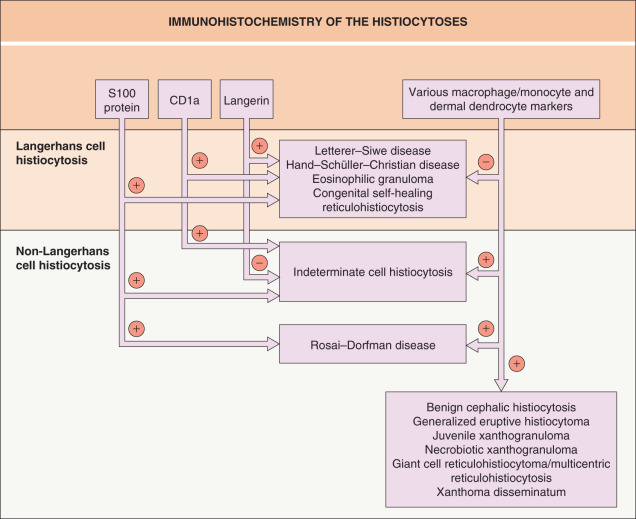
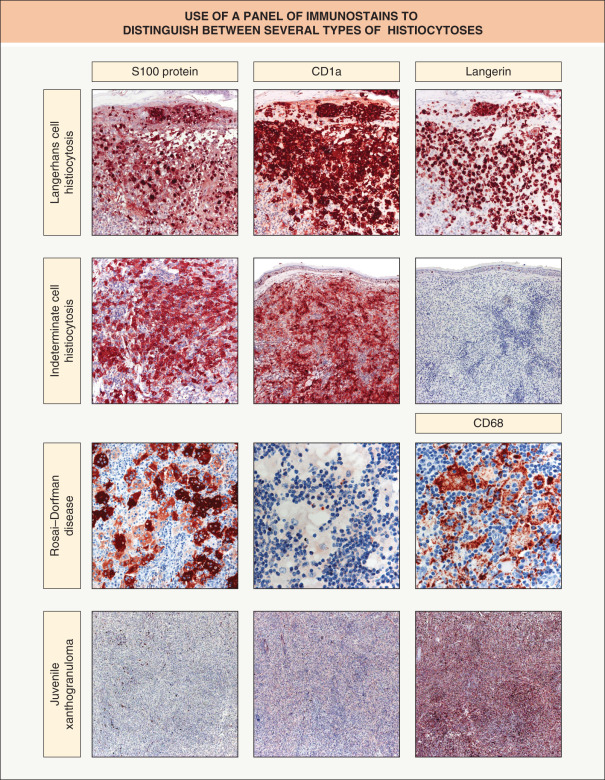
Langerhans cells are of bone marrow origin (see Ch. 4 ). Clonal LCH cells have a similar though not identical immunophenotype to Langerhans cells. Like Langerhans cells, LCH cells show positive immunostaining for S100 protein, CD1a, and Langerin (CD207) ( Table 91.2 ), and do not characteristically express factor XIIIa (a marker for type 1 dermal dendrocytes) or classic macrophage/monocyte markers such as CD68, CD163, or HAM56 ( Fig. 91.4 ). In contrast with normal Langerhans cells, LCH cells express fascin . ATPase, peanut lectin, and α-D-mannosidase are positive in LCH cells, but staining for these is less often performed, as CD1a staining is more specific. Langerin, a transmembrane C-type lectin that serves as an endocytic receptor, is a highly specific Langerhans cell marker. Because Langerin is the major protein of Birbeck granules, Langerin immunostaining can be used as a substitute for demonstration of Birbeck granules by electron microscopy. Both Langerin and CD1a have important roles in the uptake, processing and presentation of non-peptide (e.g. lipid) antigens by Langerhans cells.
| ANTIGENIC MARKERS OF THE HISTIOCYTOSES | |||||
|---|---|---|---|---|---|
| Histiocytic disorder | S100protein | CD1a | Langerin (CD207) | CD68/CD163 (KP1/KiMP or PGM1) | Factor XIIIa † |
| Langerhans cell histiocytoses | + | + | + | − | − |
| Juvenile xanthogranuloma | − (+) | − | − | + | + |
| Benign cephalic histiocytosis | − | − | − | + | + |
| Giant cell reticulohistiocytoma | − | − | − | + | + |
| Generalized eruptive histiocytoma | − | − | − | + | + |
| Indeterminate cell histiocytosis | + | + | − | + | +/− |
| Papular xanthoma ‡ | − | − | − | + | + |
| Progressive nodular histiocytosis ‡ | − | − | − | + | + |
| Hereditary ‡ progressive mucinous histiocytosis | − | − | − | + | + |
| Necrobiotic xanthogranuloma | − | − | − | + | − |
| Multicentric reticulohistiocytosis | − | − | − | + | − (+) |
| Rosai–Dorfman disease | + | − | − | + | − (+) |
| Xanthoma disseminatum | − | − | − | + | + |
† Classic dermal dendrocyte marker.
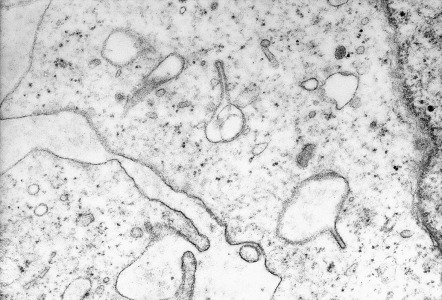
Electron microscopy demonstrates Birbeck granules, which are rod- or racquet-shaped cytoplasmic structures pathognomonic for Langerhans cells and LCH cells ( Fig. 91.5 ). On average, approximately 50% of cells will demonstrate Birbeck granules. With the availability of CD1a and Langerin staining, electron microscopy is performed less frequently today than in the past.
Differential diagnosis
The clinical differential diagnosis is vast and includes seborrheic dermatitis, various forms of diaper dermatitis or intertrigo (see Figs 13.3 and 13.11 ), and arthropod bites (including scabies), as well as leukemia, B- and T-cell lymphomas, multiple myeloma (bony lesions), urticaria pigmentosa, and the non-Langerhans cell histiocytoses (see Table 91.1 ). However, the characteristic histologic features of LCH will lead one to suspect the diagnosis, which is confirmed with the combination of positive immunostaining for CD1a, S100 protein and Langerin (CD207; Fig. 91.6 ) or demonstration of Birbeck granules on electron microscopy. Occasionally, dense dermal infiltrates of Langerhans cells can be seen in insect bite reactions, including scabies, and lead to misdiagnosis (see Fig. 85.3 ).
Treatment
All patients diagnosed with LCH should undergo evaluation of the hematologic, pulmonary, hepatosplenic, renal and skeletal systems to determine the extent of disease. Further evaluation of the CNS and the bone marrow may be required. Treatment of LCH is dependent on the number of body systems involved and the severity of involvement.
For mild, single-system skin disease (if treatment is required), topical agents, including corticosteroids, antimicrobials, mechlorethamine (nitrogen mustard) and imiquimod, and phototherapy (narrowband UVB, PUVA) have been reported to be effective in case series. For more extensive cutaneous disease, based upon case reports, thalidomide, azathioprine or methotrexate may be effective. There are also reports of the selective BRAF inhibitor vemurafenib leading to significant and rapid improvement of cutaneous disease in patients whose LCH harbored the BRAF V600E mutation. However, this drug is not viewed as first-line therapy for skin-limited disease .
Per the Histiocyte Society Evaluation and Treatment Guidelines ( www.histiocytesociety.org ), systemic therapy is recommended for patients with: (1) multisystem LCH; (2) single-system LCH with multifocal bone lesions; (3) single-system LCH with “special site” lesions, e.g. vertebral lesions with intraspinal extensions, craniofacial bone lesions with soft tissue extension; and (4) single-system LCH with at-risk CNS lesions. The LCH-III trial confirmed that a combination of vinblastine plus prednisone is effective therapy for multisystem LCH and it represents the standard regimen for multisystem LCH, with or without involvement of risk organs (see above). For those with risk organ involvement, an extended treatment regimen (12 vs 6 months) led to a lower risk of disease reactivation.
Second-line therapy consists of 2-chlorodeoxyadenosine (2-CdA) plus cytarabine (Ara-C), allogeneic hematopoietic stem cell transplantation, and in patients with BRAF V600E mutations who tend to have more aggressive disease, selective inhibitors (e.g. dabrafenib, vemurafenib). Updated information regarding ongoing clinical trials is available at www.clinicaltrials.gov and www.histiocytesociety.org .
Non-Langerhans Cell Histiocytoses
▪ Non-X histiocytosis ▪ Class II histiocytosis ▪ Histiocytoses of mononuclear phagocytes other than Langerhans cells
Benign Cephalic Histiocytosis
▪ Infantile histiocytosis with intracytoplasmic worm-like bodies
- ▪
Infants <1 year of age most commonly affected
- ▪
Red to brown macules and papules of the face and neck
- ▪
Self-limiting disease, so no treatment required
- ▪
Ultrastructural finding: worm-like bodies
Introduction
Benign cephalic histiocytosis is a rare, self-limited histiocytic proliferative disorder of young children, primarily affecting the face.
History
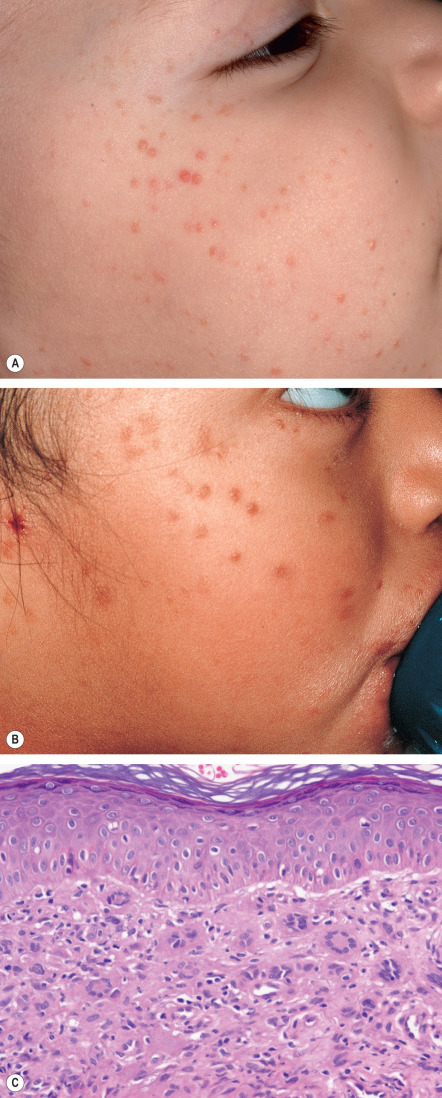
Gianotti et al. , in 1971, described the condition as “infantile histiocytosis with intracytoplasmic worm-like bodies”, based upon findings of comma-shaped structures by ultrastructural studies. When it became clear that several histiocytic disorders (e.g. LCH, juvenile xanthogranuloma) had similar ultrastructural findings, the disorder was renamed “benign cephalic histiocytosis”, based upon typical clinical findings.
Epidemiology
Benign cephalic histiocytosis is rare, with ~60 cases having been described to date. The disorder typically begins by the age of 1 year and always within the first 3 years of life. There may be a male predominance .
Pathogenesis
The pathogenesis of benign cephalic histiocytosis is not known. However, given a similar histopathology, ultrastructural appearance, and immunohistochemical profile to juvenile xanthogranuloma and generalized eruptive histiocytoma, several investigators have suggested that these three non-LCH self-healing histiocytoses should be viewed as a single disease with a spectrum of clinical features .
Clinical features
The eruption of benign cephalic histiocytosis is characterized by the evolution of 2–5 mm, pink–red to red–brown macules and papules, initially on the face ( Fig. 91.7A,B ), with subsequent appearance on the ears and neck. Occasionally, lesions may develop on the trunk and arms, with infrequent involvement of the buttocks and thighs. The papules eventually flatten, often leaving residual hyperpigmentation that fades over time. Spontaneous resolution occurs over a period of months to years. Most children are otherwise healthy without involvement of the mucous membranes or internal organs. However, diabetes insipidus has been reported in a young girl with benign cephalic histiocytosis . The course can be marked by exacerbations.