Abstract
Morphea and lichen sclerosus are inflammatory skin diseases that lead to cutaneous sclerosis, and in a number of patients, significant morbidity. The underlying pathogenesis is a combination of vascular damage, infiltration of T cells that release IL-4, and TGF-β-activated fibroblasts producing altered collagen. Morphea can be subdivided into several subgroups: plaque-type (the most common), linear (including en coup de sabre ), guttate, deep, nodular (keloidal), bullous, pansclerotic (children), and generalized. A variant of the linear morphea is Parry–Romberg syndrome in which there is progressive hemifacial atrophy. While the disability associated with morphea usually results from extension of the sclerosis into the subcutaneous tissues or over joints, the more superficial fibrosis in lichen sclerosus is most debilitating when there is genital involvement. Superpotent topical corticosteroids represent the treatment of choice for anogenital lichen sclerosus. For lichen sclerosus in other sites and the various forms of morphea, therapeutic modalities are limited and include phototherapy (UVA1, PUVA), pulse systemic corticosteroids and methotrexate.
Keywords
morphea, localized scleroderma, lichen sclerosus, plaque-type morphea, linear morphea, en coup de sabre , generalized morphea, disabling pansclerotic morphea of children, Parry–Romberg syndrome, lichen sclerosus et atrophicus, balanitis xerotica obliterans
Morphea and lichen sclerosus are inflammatory skin diseases that ultimately evolve into two distinct modes of cutaneous sclerosis. In morphea, there is inflammation of the dermis that may extend into subcutaneous structures. Lichen sclerosus is primarily a disease of the genital mucosa (and less often extragenital sites) that affects the epidermis and the dermis. Although morphea may lead to joint contractures and atrophy of underlying muscle, in general there is no involvement of internal organs. The exception is the ocular or neurologic symptoms seen in a small minority of patients with linear morphea of the head or Parry–Romberg syndrome. While some patients may develop both disorders simultaneously, they are addressed separately because of their distinct features. Therapy of these two diseases will be discussed together.
Morphea
▪ Localized scleroderma ▪ Circumscribed scleroderma ▪ Plaque-type morphea ▪ Linear morphea – linear scleroderma ▪ Morphea en coup de sabre – scleroderma en coup de sabre ▪ Deep morphea – morphea profunda
- ▪
Asymmetric sclerotic plaques, usually 2–15 cm in diameter
- ▪
Active lesions can have a lilac border with central hypo- or dyspigmentation, while inactive lesions often become hyperpigmented
- ▪
The sclerosis may extend deeply into the fat or underlying structures (e.g. fascia, muscle, bone), causing disability
- ▪
With the rare exception of ocular or neurologic symptoms in patients with linear morphea of the head, associated systemic disease is not observed
- ▪
Often progresses for several years, then regresses; the linear subtype is usually persistent
Introduction
Morphea is a clinically distinct inflammatory disease, primarily of the dermis and subcutaneous fat, which ultimately leads to a scar-like sclerosis. The small vessel changes, inflammatory infiltrate, and ultimate structural modifications are identical in morphea and systemic sclerosis (SSc), but the two diseases are distinct entities that can easily be distinguished clinically. Morphea has an asymmetric patchy or linear distribution. In contrast, SSc begins either as symmetric tightening of the fingers and hands that extends progressively toward the forearms ( limited cutaneous SSc [lcSSc]) or as symmetrically extending sclerosis of the trunk and proximal upper extremities ( diffuse cutaneous SSc [dcSSc]). Rarely, generalized morphea may resemble early dcSSc. The presence of Raynaud phenomenon, digital sclerosis, and involvement of the gastrointestinal tract and the lung in dcSSc generally allows differentiation from morphea. Distinguishing between deep morphea and eosinophilic fasciitis can prove more difficult.
Morphea can cause significant morbidity as a result of pain, skin tightness or impaired joint mobility. In ~10% of patients with morphea, sclerosis leads to significant contractures, growth retardation and even micromelia, resulting in long-term functional disability .
History
A disease with thickened skin was first mentioned by Hippocrates (400 BC). The term “scleroderma” is derived from the Greek words skleros (hard or indurated) and derma (skin). The first description of a generalized “hardness” of the skin in a young woman was by an Italian physician, Carlo Curzio, in 1753, and the French physician Gintrac coined the term “sclérodermie” in 1847. Thomas Addison has been credited with the first detailed description of morphea in 1854, which he referred to as Alibert’s keloid syndrome. In 1924, Matsui described the typical histopathologic changes of scleroderma and in 1930 O’Leary and Nomland elaborated on the distinctive features of SSc versus morphea.
Epidemiology
Even though morphea has been long recognized as a well-defined entity, few population-based studies exist. A survey from Olmsted County, Minnesota attempted to register all patients with the disorder from 1960 to 1993 . During this period, the annual incidence rate was 27 per million. According to this study, 56% of patients had plaque-type, 20% linear, 13% generalized, and 11% deep morphea .
The prevalence of morphea increases with age, occurring in approximately 500 and 2200 per million at ages 18 and 80 years, respectively . The disease is more prevalent in women than in men (2.6 : 1), with the exception of linear morphea, which has no gender preference. Data published on the incidence and prevalence of morphea probably reflect an underestimation, as they critically depend on clinical recognition and diagnosis.
Morphea is rarely life-threatening. In the Olmsted County study, the survival rate of patients with morphea was not significantly different from that of the general population . However, in 11% of the patients, substantial disability occurred. This is of particular concern since disability occurs primarily with linear morphea, which has its onset before age 18 years in two-thirds of patients .
Pathogenesis
Autoantibodies are usually as prevalent in morphea as in the general population, with two exceptions: (1) an increased prevalence of anti-single strand DNA (ssDNA), -topoisomerase IIα, -phospholipid, -fibrillin-1, and -histone antibodies (AHA) in patients with morphea (see Ch. 40 ); and (2) high titers of antinuclear antibodies (ANA) in juvenile patients with linear morphea and individuals with generalized morphea. In patients with linear morphea, the presence of ssDNA antibodies or AHA seems to be associated with increased risk of functional impairment .
Neither histology nor immunohistology of a single lesion will distinguish between morphea and SSc. In most clinicopathologic reviews, it is assumed that the two diseases are triggered by distinct events. However, the development of sclerosis following the initiating event seems to follow a common pathway. It is therefore assumed that pathogenic steps leading to SSc also contribute to the development of morphea, so they will be included in this discussion.
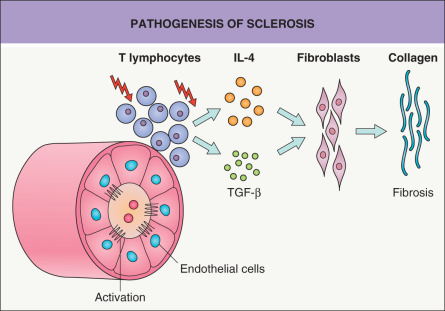
Currently, sclerosis of the skin is thought to involve three major, closely connected components: vascular damage, activated T cells, and altered connective tissue production by fibroblasts ( Fig. 44.1 ) .
Vascular Changes
A prominent feature of advanced sclerosis is a reduction in the number of capillaries. Studies performed in SSc suggest that hypoxia resulting from microvascular injury is a very early, or even the primary, event. Endothelial cells respond to various stimuli, including vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), and transforming growth factor-β (TGF-β) (see Ch. 102 ). In the sera of patients with SSc, markers indicative of endothelial cell activation (e.g. soluble adhesion molecules, VEGF) are elevated. Morphologic changes principally affect capillaries and small arterioles, and the initial changes in SSc include expression of adhesion molecules and endothelial swelling, followed by thickening of the basement membrane and intimal hyperplasia. An extrapolation is that similar changes may occur early on in morphea.
Control of Fibroblast Function by T-Cell-Derived Cytokines
Pioneering work by E Carwile Leroy showed that in vitro cultured fibroblasts isolated from sclerotic tissue produce increased amounts of collagen (types I, II and III) and other extracellular matrix proteins. These fibroblasts can maintain this phenotype for weeks over several passages. This raised the question as to whether scleroderma results from an inborn or acquired error in collagen metabolism within fibroblasts. Today, most data favor the concept that abnormal collagen production is directed by surrounding cells. T cells in particular have the capacity to modify collagen synthesis by fibroblasts, and T cells are regularly present, at least early on, in a perivascular location and especially at the leading edge of developing sclerosis (see Fig. 44.1 ).
Enhanced production of collagen and other extracellular matrix proteins is induced by T-cell-derived cytokines, especially interleukin (IL)-4, IL-13 and TGF-β. IL-4 is produced by CD4 + T helper type 2 (Th2) lymphocytes (see Ch. 4 ) and can directly enhance TGF-β production . In contrast, production of collagen and other extracellular matrix proteins can be significantly suppressed by interferons that are associated with Th1 lymphocytes . Th1 differentiation also implicates IL-12 signaling via the transcription factor STAT4. Of note, an association between STAT4 polymorphisms (e.g. rs7574865) and susceptibility to SSc, especially the limited subtype, has been reported . Additional susceptibility loci associated with SSc include immune-related genes (e.g. CD247 , IRF5, HLA region) and the promoter region of the gene that encodes connective-tissue growth factor (CTGF) .
More recently, circulating levels of the chemokine ligand CXCL4 (secreted by plasmacytoid dendritic cells) were found to be significantly elevated in SSc . Increased CXCL4 levels correlated with a higher prevalence of lung fibrosis and faster progression of skin fibrosis . Interestingly, CXCL4 promotes the Th2 cytokines IL-4 and IL-13 and has anti-angiogenic properties.
IL-4 is the most potent force in driving Th2-cell differentiation (see Fig. 4.10 ) . Because IL-4 enhances pathologic collagen production by fibroblasts and induces recruitment of eosinophils, it is believed that immune responses dominated by IL-4- as well as IL-13- and TGF-β-producing cells are critical in the initiation of skin sclerosis. This concept is supported by clinical and experimental data:
- •
In situ analysis of the advancing inflammatory margins of skin sclerosis revealed predominantly IL-4 expression.
- •
Th2-associated IL-13 and IL-33 have been shown to promote skin fibrosis in mice, and early treatment of scleroderma-developing mice with anti-IL-4 antibodies prevented scleroderma.
- •
Systemic retinoids (which interact with TGF-β signaling) appear to reduce pathologic collagen synthesis by fibroblasts and may improve skin sclerosis in patients with chronic GVHD .
- •
Th2-promoting CXCL4 is so closely associated with SSc that it has been suggested as a biomarker for SSc .
Lastly, when scleroderma fibroblasts are compared to control fibroblasts, there are differences in intracellular signaling proteins. Examples include activation of p38 mitogen-activated protein kinase and higher levels of Ha-Ras protein and reactive oxygen species. These findings plus data from animal studies demonstrating a link between skin fibrosis and pro-oxidative agents support hypoxia and oxidative stress as key factors in the pathogenesis of skin sclerosis.
Animal Model and Genetics
Currently, the best animal model for studying pathogenic events in scleroderma is the tight skin (TSK) mouse ( Fig. 44.2 ). A partial duplication of the fibrillin-1 gene may be responsible for the increased synthesis and accumulation of collagen in the skin and internal organs of TSK mice. TSK mice not only have increased collagen deposits, but also the collagen fibers are reduced in length and the quantity of hydroxyproline is increased in the dermis. Moreover, the mice have high antibody titers against topoisomerase I and fibrillin-1, similar to patients with SSc and morphea, respectively. One major difference is that blood vessels remain uninvolved in TSK mice, demonstrating that sclerosis of the skin can develop in the presence of apparently normal vessels . The phenotype can be transferred from diseased animals to healthy syngeneic mice via bone marrow cells (see Fig. 44.2 ).
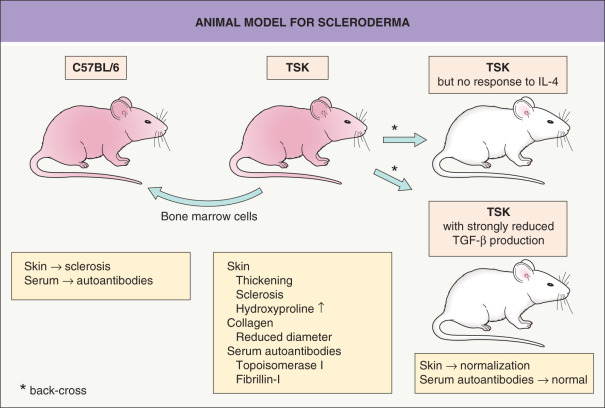
Fibroblasts from TSK mice have elevated IL-4 receptor α expression and back-crossing the TSK mice onto a genetic background that either cannot respond to IL-4 or is deficient in producing TGF-β prevents skin sclerosis. Moreover, this back-cross normalizes collagen length, skin thickness and hydroxyproline content and prevents antibody formation to topoisomerase I (see Fig. 44.2 ) . In this murine model of SSc, interventions that promoted Th1 responses or targeted PDGF receptor activity (e.g. imatinib) reduced dermal thickening and fibrosis . More recent strategies for improving experimental skin fibrosis have focused on the role of platelet-derived serotonin , β integrins, TGF-β , and Janus kinases .
Besides the phenotypic similarities, the genetic locus associated with the TSK phenotype appears to be related to the risk of developing scleroderma. In humans, this susceptibility locus is on chromosome 15q in a region that contains the fibrillin-1 gene, which is implicated in stiff skin syndrome .
Triggering Events
Most immunologic and metabolic studies have been directed toward understanding SSc, and the question remains as to whether they can also explain focal, asymmetric inflammation and sclerosis of the skin. The major discriminating factor between morphea and SSc might be the triggering event, with morphea being caused by a local trigger within the skin and SSc initiated by a systemic insult. The putative local triggering events for morphea include mechanical trauma, injections, vaccinations , and X-irradiation.
These questions initiated the search for potential triggers and studies of diseases that share clinical features with morphea and scleroderma. One intensively investigated area was the potential role of infection with Borrelia burgdorferi . The isolation of B. burgdorferi from the skin of some selected patients with morphea originally supported this speculation. However, larger studies failed to confirm this initial observation . Thus, this theory has been abandoned.
Clinical Features
Morphea can be subdivided into several subgroups: plaque-type morphea, linear morphea, generalized morphea, and other less common variants (e.g. deep, guttate, nodular). There are also inflammatory syndromes leading to superficial or deep sclerosis of the skin that resemble morphea (i.e. morpheaform; see below). Patients with morphea do not have Raynaud phenomenon or involvement of internal organs, except for underlying fascia, muscle and bone in some patients with linear morphea and the occasional patient with linear morphea of the head who has ocular or neurologic symptoms. In rare patients with presumptive morphea, clinically insignificant fibrosis of the lung or slightly reduced esophageal motility has been detected radiographically or by scintigraphy. Clinically relevant involvement of internal organs is only observed in certain types of pseudoscleroderma (see Ch. 43 ).
Plaque-Type Morphea
Plaque-type morphea, the most frequent variant, is characterized by the insidious onset of a slightly elevated, erythematous or violaceous, slightly edematous plaque that undergoes centrifugal expansion ( Fig. 44.3 ). It is generally asymptomatic and therefore frequently goes unnoticed by the patient. The central portion of the progressing lesion starts to transform into sclerotic, scar-like tissue. Depending on the depth of the sclerosis, the skin becomes progressively indurated. Centrally, it can acquire a shiny white color, and peripherally, a violaceous or “lilac” ring ( Fig. 44.4A ). Postinflammatory hyperpigmentation often dominates over the white sclerosis as the lesions mature ( Fig. 44.4B ).
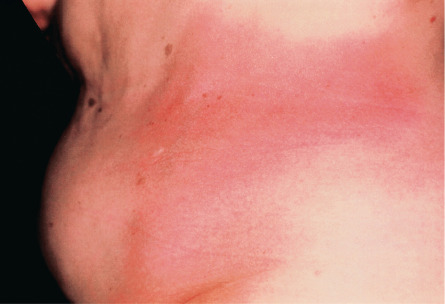
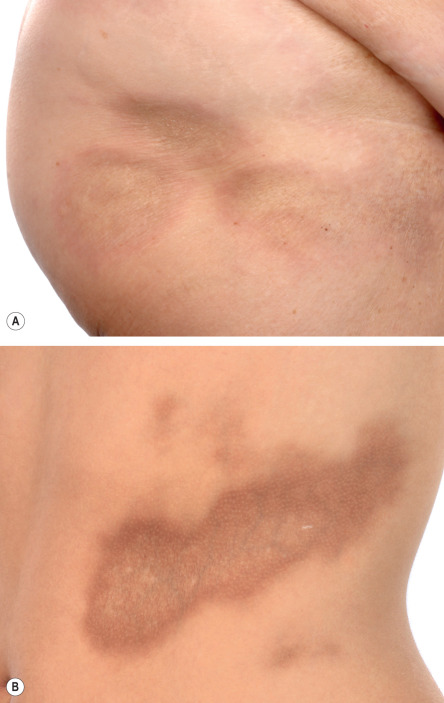
Skin structures such as hairs and sweat glands are frequently lost. Some patients complain of itch, perhaps due to associated xerosis. Once the “lilac” ring vanishes, the lesion’s progression also halts. Although this inflammatory margin may be difficult to appreciate, especially in linear sclerosis, it is an important indicator of clinical activity.
The plaques of morphea most commonly develop on the trunk and in general are between 2 and 15 cm in diameter. They are usually multiple and asymmetric, but marked variation exists. Individual lesions may enlarge significantly or remain stable in size.
The course of morphea is also variable. In most patients, morphea progresses over 3–5 years, then arrests and eventually resolves spontaneously. However, residual atrophy and dyspigmentation are commonly observed. Patients rarely have relapsing disease for more than 5 years.
Variants
Various manifestations of morphea have become associated with specific names. They do not really reflect unique entities, but rather different morphologic features, distribution patterns or depth of involvement. In most patients, morphea seems to be randomly distributed. However, in some patients, the lesions are unilateral, and they may follow dermatomes or the lines of Blaschko.
- •
Guttate morphea presents primarily as multiple, rather superficial, nummular plaques. Even though relatively small, they may become deeply indurated.
- •
Atrophoderma of Pasini and Pierini is considered by some to be a very superficial variant of plaque-type morphea, while others consider it to be a separate entity in the differential diagnosis of “burnt-out” morphea (see Ch. 99 ). Hyperpigmented patches are seen most commonly on the posterior trunk; occasionally, lesions follow the lines of Blaschko (linear atrophoderma of Moulin).
- •
Deep morphea denotes inflammation and sclerosis that affects primarily the deep dermis and subcutaneous fat and may even involve underlying structures (e.g. the fascia) ( Fig. 44.5A ). Patients normally develop a small number of hard deep plaques that may impair motility of the skin; ultimately, lesions may calcify, leading to dystrophic calcinosis cutis. Because of the location of the sclerosis, individual lesions may resemble eosinophilic fasciitis ( Fig. 44.5B and see Ch. 43 ).
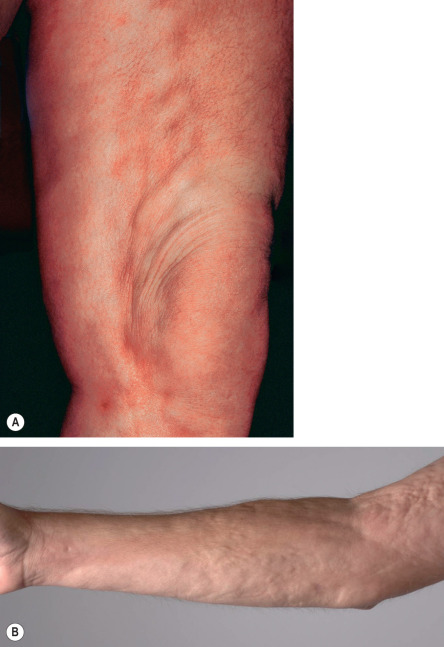
Fig. 44.5
Comparison of deep morphea and eosinophilic fasciitis.
A Note the “pseudo-cellulite” appearance of the involved skin of the thigh in deep morphea. B In eosinophilic fasciitis, the level of fibrosis is also deep, resulting in a similar rippled appearance. This patient had the eosinophilic fasciitis-like form of chronic GVHD, also referred to as chronic GVHD-related fasciitis.
B, Courtesy, Edward Cowen, MD.
- •
In nodular or keloidal morphea , inflammation within the dermis leads to thick, keloid-like nodules ( Fig. 44.6 ) or bands. It can be clinically indistinguishable from indurated keloids.
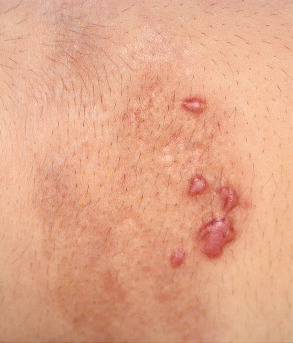
Fig. 44.6
Nodular (keloidal) morphea.
Elevated, firm pink papulonodules arising within an area of hyperpigmented induration.
Courtesy, Jean L Bolognia, MD.
- •
In rare patients, especially those where sclerosis of the skin is associated with diffuse, rapidly progressive edema, lymphoceles may result from stasis of lymphatic fluid. When the latter develop into bullae, this is referred to as bullous morphea . It is more frequently observed in generalized morphea and sclerodermoid or morpheaform GVHD and is rarely a feature of plaque-type morphea. Bullous morphea has to be distinguished from mechanical blisters that may occur centrally within plaques and are secondary to impaired mechanical stability of the dermal–epidermal junction.
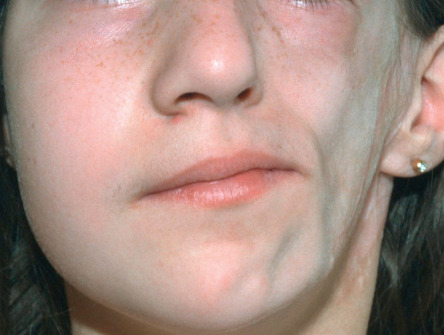
Linear Morphea and Parry–Romberg Syndrome
Linear morphea is different from plaque morphea, with respect to age of onset, distribution, clinical outcome and serologies. Morphea en coup de sabre (meaning stroke or blow with a single-edged sword) is a term used for linear morphea of the forehead and scalp. Hemifacial atrophy, or Parry–Romberg syndrome, is probably a very severe variant of linear morphea, but it may be a phenotype for more than one entity. There is a progressive loss of subcutaneous fat, but little or no sclerosis ( Fig. 44.7 ). The entire distribution of the trigeminal nerve, including the eye and the tongue, may be affected.
Linear morphea may present initially as a linear, erythematous (inflammatory) streak, but more frequently begins as a harmless-appearing lesion of plaque-type morphea that extends longitudinally as a series of plaques that then join to form a scar-like band ( Fig. 44.8 ). This band may severely impair the mobility of the affected limb. Linear morphea tends to involve the underlying fascia, muscle and tendons. This leads not only to muscle weakness but also to shortening of the muscles and fascia that impairs joint mobility. Linear morphea is especially problematic when it extends over joints, as this almost invariably results in a decrease in range of motion and functional impairment . In some patients, involvement is circular rather than linear and results in progressive atrophy of the limb similar to the Parry–Romberg variant of facial morphea. The linear variant can have prolonged periods of quiescence followed by reactivation.
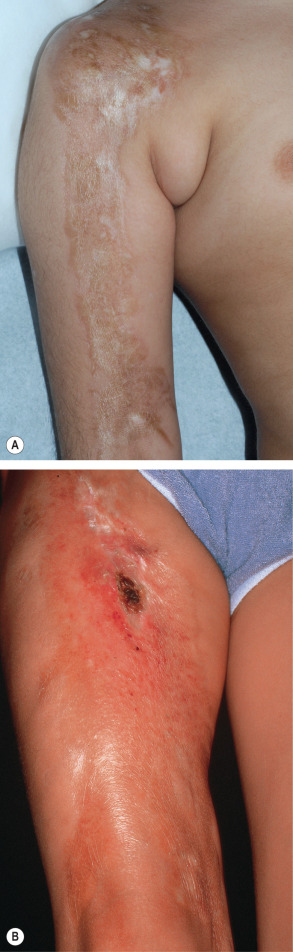
The en coup de sabre type of morphea represents linear morphea of the head ( Fig. 44.9 ). It is typically unilateral and extends from the forehead into the frontal scalp. It may start either as a linear streak, which may initially mimic a port-wine stain, or as a row of small plaques that coalesce. A paramedian location is most common. Like plaque morphea, it may initially be surrounded by a discrete lilac ring that extends longitudinally and may reach the eyebrows, nose and even the cheeks. As inflammation wanes, a linear, hairless crevice develops that in some patients is sclerotic, while in others appears more atrophic.
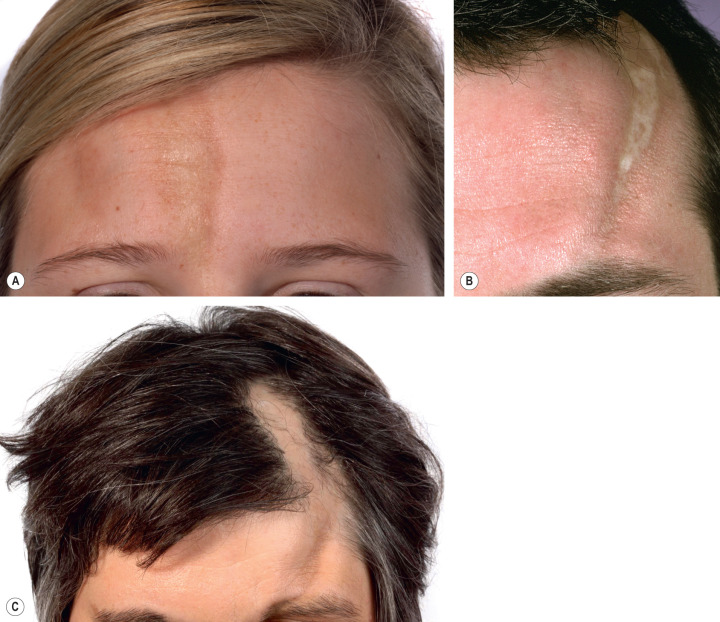
En coup de sabre morphea can also involve the underlying muscles and osseous structures. Rarely, the inflammation and sclerosis progress to involve the meninges and even the brain, creating a potential focus for seizures .
Generalized Morphea
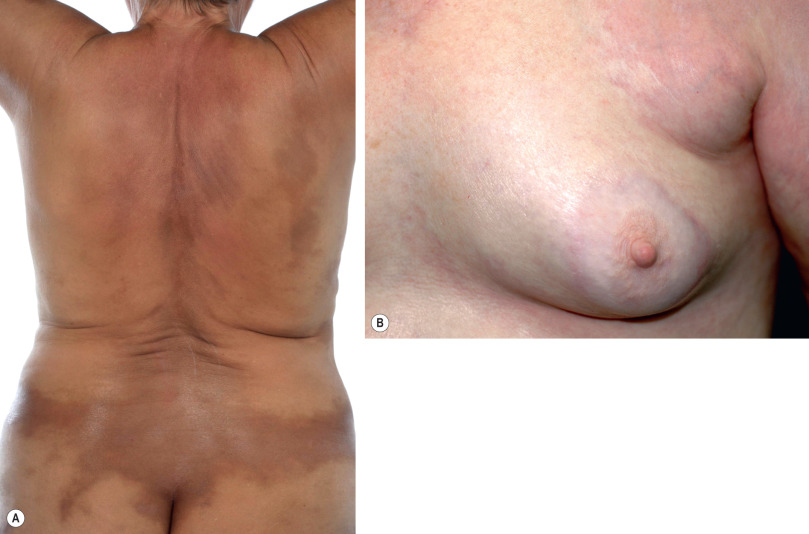
Generalized morphea normally begins insidiously on the trunk as plaque morphea. An individual lesion is indistinguishable from classic plaque morphea, except that it does not stop expanding. Just as in the diffuse form of SSc, multiple plaques rapidly coalesce and can affect nearly the entire trunk, often only sparing the nipples ( Fig. 44.10 ). The sclerosis may involve the extremities down to the hands (presenting initially as puffy edema). As the cutaneous sclerosis progresses, it may result in disabling constrictions that even cause difficulty in breathing due to impaired thorax mobility and inflammation of the intercostal muscles. Although an aggressive therapeutic approach is recommended, the disease is usually persistent given its often limited response to treatment.
Morphea in Childhood
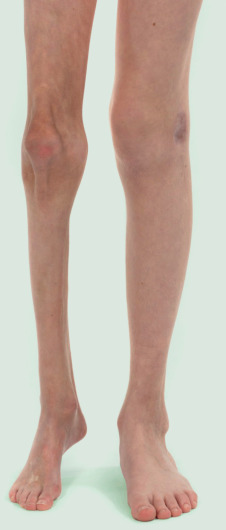
Approximately 20% of individuals with morphea are children and teenagers. The female-to-male ratio for plaque-type morphea in this age group is about 2 : 1, with a mean age at disease onset of 7 years . In two-thirds of patients with linear morphea, disease onset is prior to age 18 years, providing an explanation for the possibility of growth retardation of the affected limb. Thus, if left untreated, linear morphea may result not only in decreased range of motion of joints but also in permanent limb asymmetry from unilateral hypoplasia ( Fig. 44.11 ).
Disabling pansclerotic morphea of children is similar to generalized morphea of the adult. It usually starts before 14 years of age and tends to cause lifelong severe disability due to persistent atrophy of the underlying muscles and contractures of the involved joints. The disease tends to extend from the trunk to the hands and feet. Periodontal atrophy may occur, but there is only slight involvement of the esophagus and lung. Distinction from SSc may prove challenging.
Importantly, in a large multicenter study of children with morphea, 22% had extracutaneous findings that were primarily articular (11%), neurologic (4%), and ocular (2%) . The latter two findings were seen primarily in those with linear morphea of the head or Parry–Romberg syndrome, i.e., 85% of affected patients had these two variants.
Laboratory Findings
Laboratory abnormalities are not a prominent feature of morphea, except for generalized and linear morphea. The ESR and serum protein levels are usually normal, but eosinophilia may occur, especially during early active phases of the disease. High titers of ANA or antibodies to ssDNA and histones can be detected in 40–80% of patients with linear and generalized morphea, but are uncommon in adults with plaque-type morphea (see above). Approximately 40% of children and adolescents with morphea have elevated ANA titers .
Pathology
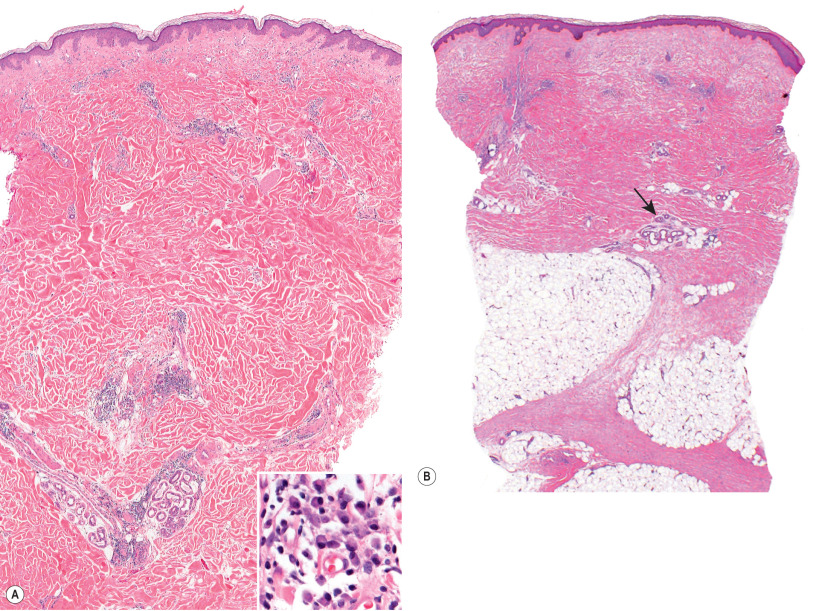
The histology of morphea depends on two factors: the stage and area of the lesion sampled (inflammatory margin or central sclerosis) and the depth to which the disease extends. In most situations, morphologic changes are best seen at the border between the dermis and subcutaneous fat. Specimens for histology must include subcutaneous fat and it is important to note whether the biopsy specimen is taken from the inflammatory border or from the fibrotic center.
At the inflammatory border, vascular changes are relatively discrete by light microscopy. Vessel walls show endothelial swelling and edema. Capillaries and small arterioles are surrounded by an infiltrate that contains primarily CD4 + T cells and sometimes eosinophils, plasma cells and mast cells ( Fig. 44.12A ).