Abstract
Smooth muscle, adipose and cartilage neoplasms arising in the skin and subcutis comprise a wide spectrum of benign and malignant neoplasms that show myocyte, adipocyte, and chondrocyte differentiation, respectively. Tumors of fat, especially lipomas, occur most commonly. These neoplasms often present as solitary lesions with significant overlap in clinical appearance. As with other cutaneous tumors, multiple lesions raise the possibility of an inherited disease (e.g. hereditary leiomyomatosis and renal cell cancer). The histopathologic diagnosis is established primarily on the basis of characteristic morphologic features, including architecture and cytomorphology, as well as identification of the predominant direction and level of differentiation. For several of these neoplasms, immunohistochemical studies provide helpful diagnostic information. Cytogenetic analysis and molecular studies are increasingly being used for diagnosis and classification of certain tumors based upon relatively histiotype-specific karyotypic abnormalities. The treatment of choice for solitary, benign (if requested) or relatively non-aggressive malignant neoplasms is excisional surgery with clear margins.
Keywords
leiomyoma, cutaneous leiomyoma, piloleiomyoma, leiomyosarcoma, subcutaneous leiomyosarcoma, dermal leiomyosarcoma, smooth muscle hamartoma, lipoma, angiolipoma, spindle cell lipoma, pleomorphic lipoma, hibernoma, nevus lipomatosus superficialis, lipoblastoma, lipoblastomatosis, liposarcoma, atypical lipomatous tumor, extraskeletal chondroma, hereditary leiomyomatosis and renal cell cancer
Introduction
Smooth muscle, adipose and cartilage neoplasms arising in the skin and subcutis comprise a wide spectrum of benign and malignant neoplasms that show myocyte, adipocyte, and chondrocyte differentiation, respectively. Tumors of fat, especially lipomas, occur most commonly. These neoplasms often present as solitary lesions with significant overlap in clinical appearance. In addition, the clinical differential diagnosis can be rather broad ( Fig. 117.1 ). As with other cutaneous tumors, multiple lesions raise the possibility of an inherited disease (e.g. hereditary leiomyomatosis and renal cell cancer).
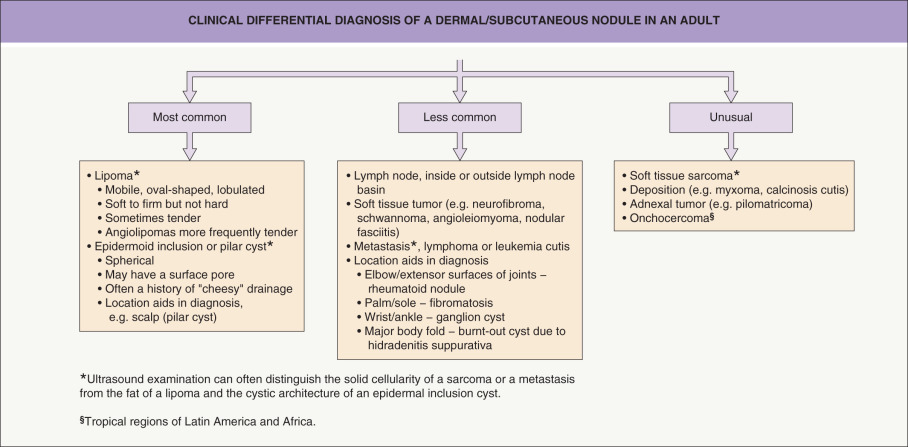
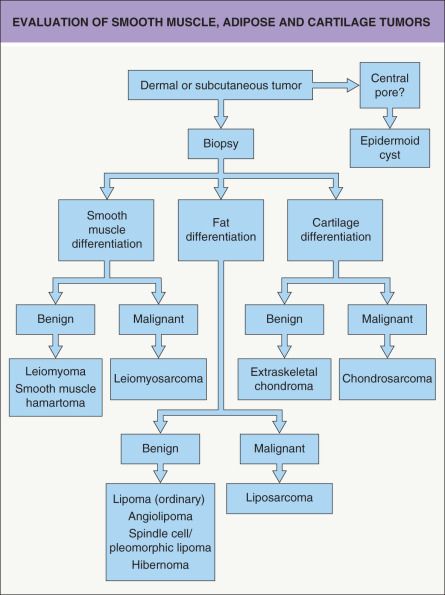
In this group of neoplasms, the histopathologic diagnosis is established primarily on the basis of characteristic morphologic features, including architecture and cytomorphology, as well as identification of the predominant direction and level of differentiation ( Fig. 117.2 ). Determination of whether a tumor is benign or malignant also requires assessment of features such as circumscription and if atypia and increased mitotic activity are present. Great care is necessary when applying traditional morphologic criteria for malignancy to cutaneous mesenchymal neoplasms because some clinically less aggressive tumors may exhibit prominent atypical features. In other words, aggressive behavior is not always predictable based solely on morphologic findings. For several of these neoplasms, immunohistochemical studies provide helpful diagnostic information ( Table 117.1 ). Cytogenetic analysis and molecular studies are increasingly being used for diagnosis and classification of adipose tumors based upon relatively histiotype-specific karyotypic abnormalities (see below).
| IMMUNOHISTOCHEMISTRY OF SELECTED SMOOTH MUSCLE, ADIPOSE AND CARTILAGE NEOPLASMS | ||
|---|---|---|
| Neoplasm | Cell type(s) | Stain(s) |
| Leiomyoma | Myocytes |
|
| Leiomyosarcoma | Myocytes |
|
| Lipoma | Adipocytes |
|
| Angiolipoma | Adipocytes, endothelial cells |
|
| Spindle cell/pleomorphic lipoma | Spindle and multinucleated giant cells, adipocytes |
|
| Hibernoma | Adipocytes, spindle cells |
|
| Lipoblastoma/lipoblastomatosis | Adipocytes |
|
| Liposarcoma/atypical lipomatous tumor | Adipocytes, adipoblasts |
|
| Extraskeletal chondroma | Chondrocytes |
|
A number of tumor-related and patient-related factors may influence the choice of treatment, including tumor type and size, anatomic location, patient’s life expectancy, presence of comorbidities, and cosmetic concerns. For the majority of solitary, benign or relatively non-aggressive malignant neoplasms, excisional surgery with clear margins is the treatment of choice. However, removal of a benign tumor is often done at the patient’s request rather than for medical reasons. Other modalities, e.g. cryotherapy, laser ablation (multiple leiomyomas), liposuction (large lipomas), may be preferable in certain patients. For more aggressive malignant tumors, the therapeutic goal is complete eradication, via either wide local excision or Mohs micrographic surgery, while trying to preserve normal function and avoiding mutilating surgery. As expected, the most significant predictor of recurrences is margin status. In some dermal sarcomas, extension into the subcutis represents a negative prognostic factor.
Tumors of Smooth Muscle
Leiomyoma
▪ Superficial leiomyoma ▪ Leiomyoma cutis ▪ Superficial benign smooth muscle tumor
- ▪
Three distinct variants: piloleiomyoma, genital leiomyoma, and angioleiomyoma
- ▪
A solitary or multiple clustered (piloleiomyomas) papules or nodules in an adult
- ▪
Piloleiomyomas and angioleiomyomas may be painful
- ▪
Multiple piloleiomyomas may be a manifestation of hereditary leiomyomatosis and renal cell cancer (Reed syndrome)
- ▪
Smooth muscle cells (myocytes) are fusiform cells with centrally located, cigar-shaped nuclei
Introduction
Superficial (cutaneous) leiomyomas represent benign smooth muscle tumors that can arise from: (1) arrector pili muscles; (2) the dartoic, vulvar or mammary smooth muscles; or (3) the muscles enveloping dermal blood vessels. Thus, they are subclassified into piloleiomyomas (solitary or multiple), leiomyomas of the external genitalia, and angioleiomyomas. Some authors categorize vulvar and scrotal leiomyomas separately, based upon distinctive clinical and histologic features .
History
In an early review article, Arthur Purdy Stout cites an 1854 publication of Rudolf Virchow as the first case report of multiple leiomyomas involving the skin near the areola in a 32-year-old man.
Epidemiology
Cutaneous leiomyomas are relatively uncommon neoplasms, but their exact incidence is not known . Angioleiomyomas appear to be more common than piloleiomyomas , with genital leiomyomas the least common variant.
Pathogenesis
The exact pathogenesis of superficial leiomyomas has yet to be determined. Piloleiomyomas are thought to arise from the smooth muscle cells within the arrector pili muscle of the folliculosebaceous unit. Leiomyomas of the external genitalia are probably derived from the network of smooth muscles in the external genitalia (e.g. scrotum, vulva) and areolae. Angioleiomyomas are believed to originate from the smooth muscle within the walls of arteries and veins.
Multiple piloleiomyomas may occur sporadically or they may be inherited in an autosomal dominant manner (with variable penetrance) as a component of hereditary leiomyomatosis and renal cell cancer (HLRCC), also referred to as Reed syndrome, in which patients have multiple cutaneous leiomyomas and uterine leiomyomatosis as well as renal cell cancer ( Table 117.2 ). Although the molecular basis of HLRCC is known (mutations in the gene that encodes fumarate hydratase), how this translates into smooth muscle tumors is not understood.
| CLINICAL FEATURES OF HEREDITARY LEIOMYOMATOSIS AND RENAL CELL CANCER (REED SYNDROME) | |
|---|---|
| Genetic aspects |
|
| Clinical features |
|
| Screening of individuals with multiple cutaneous leiomyomas |
|
| Patients with confirmed syndrome |
|
| Established diagnosis: heterozygous pathogenic variant in FH + multiple cutaneous leiomyomas (at least 1 confirmed histologically) – or – single cutaneous leiomyoma + family history of syndrome – or – ≥1 tubulo-papillary, collecting-duct, or papillary type 2 renal tumor +/− family history | |
Multifocal subcutaneous leiomyomas arising in the setting of immunodeficiency, including HIV infection, may be associated with Epstein–Barr virus infection.
Clinical features
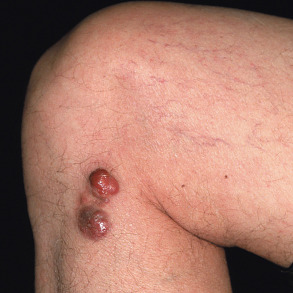
Piloleiomyomas can be solitary or multiple, and in an occasional patient may even number in the hundreds. They present as firm, reddish-brown to skin-colored papules or nodules. Solitary piloleiomyomas develop primarily during adulthood and there is an approximately equal sex distribution ; rare congenital or pediatric cases have been reported. When multiple, the distribution pattern is most often clustered ( Fig. 117.3 ), linear or along the lines of Blaschko, but widespread lesions are observed occasionally. Most piloleiomyomas measure 1–2 cm in diameter, and they usually develop on the extremities and trunk (especially the shoulder), with solitary lesions favoring the limbs and multiple lesions the trunk . Piloleiomyomas are often associated with spontaneous or induced pain, e.g. following cold exposure, but the explanation is still unclear .
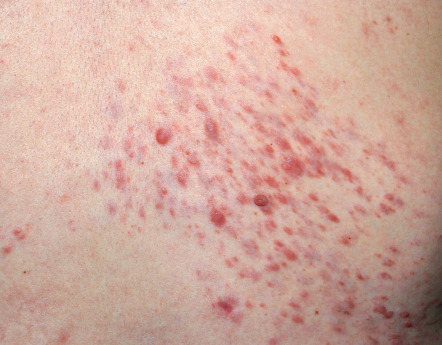
Genital leiomyomas are most often solitary and painless. They can develop on the vulva, penis, scrotum, nipple or areola and usually measure less than 2 cm in diameter. Occasionally, lesions are pedunculated.
Angioleiomyomas most commonly present as a solitary, firm subcutaneous nodule on the lower extremity. They often develop in women during the fourth to sixth decades of life . Approximately 50% of angioleiomyomas are painful.
Pathology
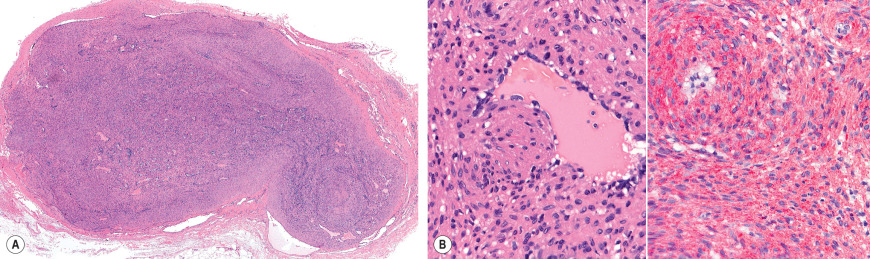
Piloleiomyomas are typically centered within the reticular dermis, with focal extension into the subcutaneous fat in some cases. They are composed of interlacing fascicles of relatively bland smooth muscle cells (myocytes), which peripherally tend to ramify between surrounding dermal collagen bundles ( Fig. 117.4 ); often there is a close relationship to hair follicles . Occasionally, the overlying epidermis is hyperplastic. Smooth muscle cells have abundant brightly eosinophilic cytoplasm and blunt-ended, cigar-shaped nuclei.
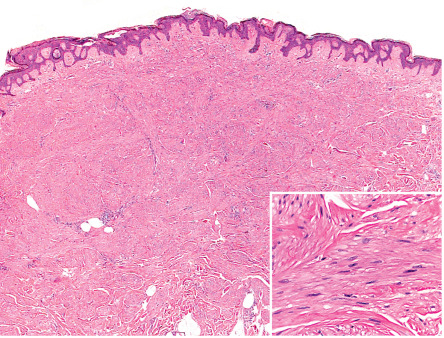
Mitotic figures are rare. In the presence of bland cytology, one or two morphologically normal mitotic figures per 10 high-power fields may be seen, which does not signify aggressive behavior. Cases with nuclear pleomorphism and atypical nuclei, but without mitotic activity (symplastic leiomyomas), have also been described .
In leiomyomas, smooth muscle differentiation is usually obvious in H&E-stained sections. Sometimes, special stains for smooth muscle cells are utilized (see Table 117.1 ). Tumor cells consistently stain positively for both smooth muscle actin (SMA) and desmin. In HLRCC, there is often negative immunohistochemical staining for fumarate hydratase in the cutaneous leiomyomas (in one series, 83% sensitivity/75% specificity ). Intracellular accumulation of S-(2-succino)-cysteine in uterine leiomyomas may suggest fumarate hydratase aberration but this is not specific for HLRCC . In contrast to uterine leiomyomas, cutaneous leiomyomas do not express receptors for estrogen or progesterone .
Genital leiomyomas of the scrotum or vulva tend to be larger and better circumscribed than piloleiomyomas. Scrotal leiomyomas are usually homogeneous spindle cell tumors, while vulvar lesions may demonstrate an epithelioid cytology, myxoid changes, and/or hyalinization . Leiomyomas of the nipple closely resemble piloleiomyomas.
Angioleiomyomas are usually well-circumscribed tumors centered within the superficial subcutis ( Fig. 117.5 ). They are composed of bland smooth muscle cells compactly arranged into bundles and whorls around thick-walled vascular channels . Degenerative changes including hyalinization, focal thrombosis, myxoid changes, dystrophic calcification, pyknotic nuclear atypia, and fat foci may be present. Tumors composed of a proliferation of blood vessels, smooth muscle cells, and adipose tissue are termed angiomyolipomas or angiolipoleiomyomas.
Differential diagnosis
For single lesions, the clinical appearance is nonspecific and the differential diagnosis includes a wide range of entities that can present as solitary dermal nodules, including dermatofibromas. When a lesion is painful or there is a clustering of multiple red–brown papulonodules, the diagnosis may be strongly suspected prior to biopsy.
Histopathologically, leiomyomas need to be distinguished from other spindle cell tumors with fibrohistiocytic or peripheral nerve sheath differentiation.
Treatment
Simple excision is usually curative for solitary or limited tumors. For those tumors that are not amenable to surgery because of their number or location, treatment with gabapentin, medications that reduce smooth muscle contraction (e.g. nifedipine, nitroglycerin, doxazosin), cryotherapy, or CO 2 laser ablation may provide relief from pain .
Leiomyosarcoma
▪ Superficial leiomyosarcoma ▪ Superficial malignant smooth muscle tumor
- ▪
Malignant smooth muscle neoplasms
- ▪
Occurs primarily in adults >50 years of age
- ▪
Most commonly located on the extremities, in particular the lower leg
- ▪
Dermal leiomyosarcomas are typically cured by simple excision and rarely metastasize; subcutaneous leiomyosarcomas require wide excision and have a 25–40% risk of metastasis
Introduction
For management and prognostic reasons, it is important to differentiate between “superficial leiomyosarcomas” that are limited to the dermis and lesions that primarily arise in or extensively involve the subcutis. The former have a favorable prognosis and can recur, but very rarely metastasize. Subcutaneous leiomyosarcomas, in contrast, metastasize in approximately 25–40% of cases .
Epidemiology
Cutaneous and subcutaneous leiomyosarcomas are rare neoplasms, comprising only 4–6.5% of soft tissue sarcomas .
Pathogenesis
Dermal leiomyosarcomas are thought to arise from either arrector pili or genital smooth muscles. Subcutaneous leiomyosarcomas probably originate from vascular smooth muscle. Anecdotal reports describe a rare association with trauma and radiation .
Clinical features
Cutaneous and subcutaneous leiomyosarcomas classically present as a solitary, firm, skin-colored to red–brown nodule or plaque. Dermal tumors range from 0.5 to 4 cm in diameter , while subcutaneous lesions tend to be larger. Sometimes the tumors are painful or become ulcerated. Leiomyosarcomas favor the extremities, in particular the lower leg of older adults ( Fig. 117.6 ) . There is a slight male preponderance . Patients with multiple tumors should be evaluated to exclude the possibility of metastases due to a primary retroperitoneal or visceral leiomyosarcoma.
Pathology
The histopathologic features of leiomyosarcomas span a morphologic continuum that, at the well-differentiated end, demonstrates an overlap with leiomyomas, while poorly differentiated lesions closely resemble atypical fibroxanthomas or “malignant fibrous histiocytomas” . Leiomyosarcomas of moderate differentiation are comprised of cells that cytologically resemble normal smooth muscle cells. However, compared to leiomyomas, malignant smooth muscle tumors are more cellular, exhibit cytologic atypia, and contain easily identifiable mitotic figures ( Fig. 117.7 ). At least focally, the spindle cells are arranged in fascicles. Cytologically, poorly differentiated leiomyosarcomas may resemble myofibroblastic or fibrohistiocytic tumors. Notably, the degree of differentiation may vary within a single tumor. Rarely, the cells have an epithelioid appearance .
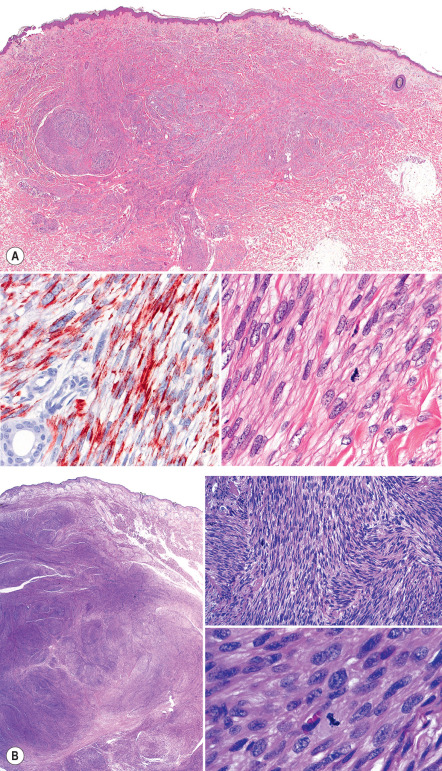
A definitive diagnosis of leiomyosarcoma often requires adjuvant special stains and/or immunohistochemical studies to support the cell lineage (see Table 117.1 ). Leiomyosarcomas consistently stain positive for SMA and desmin. Of the two markers, actin is more sensitive and desmin is somewhat more specific. By definition, dermal leiomyosarcomas are superficial malignant smooth muscle neoplasms with at least 90% of the tumor confined to the dermis . They are usually poorly circumscribed and are composed of spindle cells; the latter are focally arranged in fascicles that infiltrate between adjacent collagen bundles. In comparison, subcutaneous leiomyosarcomas tend to be better circumscribed and may be surrounded by a pseudocapsule of compressed tissue.
Differential diagnosis
Because the clinical features are not unique, the differential diagnosis includes a number of tumors that can present as solitary plaques, e.g. dermatofibrosarcoma protuberans, or firm nodules (see Fig. 117.1 ).
The histopathologic differential diagnosis usually raises two problems: the differentiation of leiomyosarcoma from leiomyoma and its delineation from other spindle cell neoplasms. Traditionally, the number of mitotic figures is used as the most reliable diagnostic criterion when trying to predict clinical behavior. In the presence of cytologic atypia, a mitotic rate of one or more mitotic figures per high-power field is generally considered to be indicative of malignancy . It is important to remember, however, that the prognosis of leiomyosarcomas appears to depend more on the depth of involvement (dermal vs subcutaneous), and the extent of mitotic activity is probably more important in subcutaneous lesions. Necrosis and angiolymphatic invasion are generally not useful in predicting prognosis for patients with dermal and subcutaneous leiomyosarcomas.
The distinction of leiomyosarcoma from other dermal spindle cell neoplasms nearly always requires immunophenotyping. This is best approached by using a panel of antibodies that examine the neoplasm for its line of differentiation ( Table 117.3 ). A standard panel consists of antibodies to actin, desmin, cytokeratin, S100 protein, and CD68. Leiomyosarcoma will usually be actin-positive and desmin-positive, but negative for keratin, S100 protein, and CD68.
| IMMUNOHISTOCHEMICAL FINDINGS IN LEIOMYOSARCOMA RELATIVE TO OTHER SPINDLE CELL TUMORS | |||||
|---|---|---|---|---|---|
| Leiomyosarcoma | SCC | Melanoma | Nerve sheath tumors | AFX/MFH | |
| Actin | + | − | − | − | −/+ |
| Desmin | +/− | − | − | − | − |
| Cytokeratin | −/+ | + | − | − | − |
| S100 protein | −/+ | − | + | + | −/+ |
| CD68 | − | − | +/− | − | +/− |
| h-Caldesmon | +/− | − | − | − | − |
| CD10 | +/− | +/− | −/+ | +/− | + |
Treatment
Wide excision with meticulous examination of all surgical margins is crucial in order to prevent recurrence. Recurrent tumors tend to infiltrate more deeply, and they are often less well differentiated. They are more difficult to treat and carry a higher risk of metastasis. Several authors have reported successful treatment with Mohs micrographic surgery , allowing for complete margin control.
Complete surgical excision of a primary dermal leiomyosarcoma can be curative. Local recurrences, due to incompletely excised tumors, are unusual and metastases rarely, if ever, develop. Recently, some authors have even suggested using the term “atypical intradermal smooth muscle neoplasm” .
Subcutaneous leiomyosarcomas metastasize in 25–40% of patients. The risk of metastasis appears to be related to size, as metastasis is unlikely in tumors <5 cm in diameter. Metastatic spread occurs hematogenously to the lung and, less often, other visceral organs, or via the lymphatics to regional lymph nodes .
Smooth Muscle Hamartoma
- ▪
Congenital or acquired skin-colored to hyperpigmented plaque on the trunk or proximal extremities
- ▪
Commonly exhibit follicular prominence and/or hypertrichosis
- ▪
Clinical and histopathologic overlap with Becker melanosis (nevus)
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


