Abstract
Panniculitis represents infiltration of the subcutaneous tissue by inflammatory cells, although the term has also been applied to neoplastic infiltration. Typically there is deep induration or swelling of the skin, accompanied by erythema, warmth, pain, and sometimes ulceration or drainage. Occasionally, induration or nodularity may be present without significant inflammation clinically or these changes may persist after inflammation has abated. There are numerous causes of panniculitis and given the overlap in clinical and pathologic features, findings are not always diagnostic of a particular entity. That said, there is a group of primary panniculitides, each of which has recognizable clinical, histopathologic and/or laboratory features that in general point to a specific diagnosis and targeted therapeutic approaches. These conditions comprise the subject of this chapter.
Keywords
panniculitis, subcutis, erythema nodosum, erythema induratum, alpha-1 antitrypsin deficiency panniculitis, connective tissue panniculitis, pancreatic panniculitis, sclerema neonatorum, subcutaneous fat necrosis of the newborn, post-steroid panniculitis, lupus panniculitis, traumatic panniculitis, lipodermatosclerosis, infection-induced panniculitis, factitial disease
Introduction
Panniculitis is a diagnostically challenging arena for dermatologists and pathologists. Terminology is difficult, partly because various names have been applied to the same disorder (e.g. nodular vasculitis and erythema induratum), and partly because new discoveries have resulted in the introduction of new terms and the abandonment of others. From a clinical standpoint, many forms of panniculitis with diverse etiologies closely resemble one another, presenting as tender, erythematous, subcutaneous nodules. Some panniculitides can be a manifestation of different disease processes (erythema nodosum is the classic example), and, even if the type of panniculitis is correctly identified, this is only the first step in a series of investigations required to determine the underlying cause. From a pathologic standpoint, the subcutaneous fat responds to a variety of different insults in a limited number of ways, and, therefore, histopathologic differences among the various forms of panniculitis may be subtle. Management can also be difficult, since there are often at least two therapeutic desiderata:
- •
specific treatment of the panniculitis
- •
treatment of the underlying illness.
In this chapter, these issues will be addressed by introducing a schema for the classification of these disorders, recommending an approach to the histopathologic diagnosis, and providing information about the specific forms of panniculitis and their management.
Table 100.1 provides a working classification for the multiple forms of panniculitis. The categories are determined partly by clinical characteristics such as anatomic location ( Fig. 100.1 ), partly by histopathology, and partly by etiology. A word should be said about septal versus lobular panniculitis. These are largely artificial constructs, since there is no purely septal or purely lobular panniculitis. Certain forms of panniculitis can be characterized as having predominantly septal involvement, and this finding can provide a useful clue to diagnosis when combined with other clinical and histopathologic features.
| CLASSIFICATION OF THE PANNICULITIDES |
|---|
| Predominantly septal panniculitis |
|
| Lobular and mixed septal–lobular panniculitis |
|
* Descriptions vary; some authors regard this as a lobular or mixed septal–lobular panniculitis.
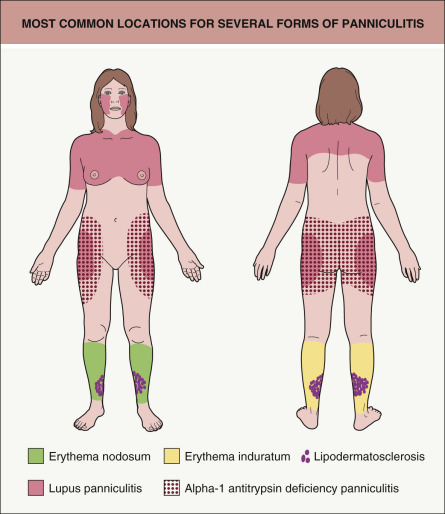
Table 100.2 provides an approach to the histopathologic diagnosis of an unknown case of panniculitis. When performing a biopsy in a patient with panniculitis, it is absolutely critical that the specimen includes a generous portion of subcutaneous fat. Therefore, excisional biopsies extending through the subcutis or narrow incisional biopsies that incorporate a broad expanse of subcutaneous fat are preferable to punch biopsies.
| AN APPROACH TO THE HISTOPATHOLOGIC DIAGNOSIS OF PANNICULITIS | |
|---|---|
| Histopathologic aspects to consider | Conclusions |
| Look for the “center of gravity” of the infiltrate | Recommended originally by Pinkus, the purpose is to determine whether or not the inflammatory process is centered in the subcutis (favoring a primary panniculitis), in the dermis, or in the fascia (in which case the panniculitis may be a secondary manifestation of a deeper inflammatory process) |
| Determine if the panniculitis is predominantly septal, predominantly lobular, or mixed | Generally, a predominantly septal panniculitis narrows the diagnostic considerations (see Table 100.1 ). If the panniculitis is lobular or mixed, look for other distinguishing features |
| Determine if there is vasculitis involving a medium-sized vessel, in the face of a lobular or mixed panniculitis | This is characteristic of erythema induratum (nodular vasculitis); caveat: deeper levels are often required in order to demonstrate the involvement of vessels |
| Look for fat necrosis with saponification and “calcium soap” formation | This is a feature of pancreatic panniculitis |
| Look for needle-shaped clefts within lipocytes | Their presence favors a diagnosis of sclerema neonatorum, subcutaneous fat necrosis of the newborn , or post-steroid panniculitis |
| Determine if the panniculitis has a lymphoplasmacytic predominance | This tends to be a feature of panniculitis due to connective tissue disease , including lupus erythematosus; beware of lymphocytic predominance in cases of subcutaneous panniculitis-like T-cell lymphoma (see Table 100.9 ) |
| Check for a “central nidus” of inflammation or evidence of a needlestick injury. Look for vacuolated spaces or foreign material | These are clues to traumatic panniculitis including factitial panniculitis . Polarization microscopy can be helpful |
| Determine if lobular panniculitis has a predominance of neutrophils | Infection-induced panniculitis, neutrophilic panniculitis associated with inflammatory bowel disease and rheumatoid arthritis, subcutaneous Sweet syndrome, traumatic panniculitis ; less so in alpha-1 antitrypsin deficiency panniculitis and pancreatic panniculitis |
| Look for membranocystic changes in the subcutis | This change is characteristic of (though not pathognomonic for) lipodermatosclerosis; it can be observed in any panniculitis with prominent degenerative changes |
| Determine if the panniculitis is associated with substantial cellular necrosis, vascular proliferation, hemorrhage, sweat gland necrosis, or neutrophilic aggregates | These changes are often encountered in infection-induced panniculitis . Tissue cultures and special stains for organisms may be indicated |
| Determine if the infiltrating cells are particularly monotonous or atypical in appearance | Consider subcutaneous panniculitis-like T-cell lymphoma and other lymphomas with involvement of the subcutaneous fat. Rarely, non-hematologic malignancies mimic panniculitis. Immunohistochemical stains are mandatory for a precise diagnosis |
| Determine if there are large cells with cytophagic activity | Cytophagic activity (hemophagocytosis) observed primarily in subcutaneous infiltrates of primary cutaneous γ/δ T-cell lymphoma. This finding was previously termed “cytophagic histiocytic panniculitis” |
Erythema Nodosum
▪ Erythema contusiformis ▪ Erythema nodosum migrans (variant form)
- ▪
Tender, erythematous, subcutaneous nodules
- ▪
Usually distributed symmetrically and favor the pretibial areas; occasionally occur elsewhere
- ▪
In later stages, lesions acquire a bruise-like appearance
- ▪
May be accompanied by fever, arthralgias and malaise
- ▪
Associated with a wide variety of systemic disorders
Introduction
Erythema nodosum is the best known of the various forms of panniculitis, as well as the most common. It typically presents as an acute eruption of tender, erythematous, subcutaneous nodules in the pretibial areas bilaterally. It is widely regarded as a delayed hypersensitivity response to a variety of antigenic challenges , although the mechanisms of its development are more complex than this statement would indicate. Histopathologically, it is the prototype of a “septal” panniculitis. Identification and treatment of the underlying disorder, if found, is of primary importance, but therapy directed toward the lesions themselves is also an option, especially when idiopathic.
History
At the beginning of the eighteenth century, Robert Willan gave the first clear description of erythema nodosum, and he provided its name in his famous work, On Cutaneous Disease .
Epidemiology
Erythema nodosum can occur at any age, in both sexes, and in all racial groups. It is more common among women and is more frequently observed during the second through fourth decades of life . The relative ranking of underlying causes may vary according to geographic location; for example, in areas where Coccidioides immitis is endemic or regions where Behçet disease is more prevalent.
Pathogenesis
Erythema nodosum has been considered a delayed hypersensitivity response to a variety of antigenic stimuli, including bacteria, viruses and chemical agents . Llorente et al. observed expression of mRNA for Th1 cytokines (interferon-γ, interleukin-2) in the skin lesions and peripheral blood of patients with erythema nodosum, and a Th1 pattern of cytokine synthesis is associated with delayed-type hypersensitivity reactions. However, a complex series of intermediate steps is involved in the development of these lesions. A variety of adhesion molecules and inflammatory mediators appear to be associated with erythema nodosum. For example, in erythema nodosum lesions, vascular cell adhesion molecule-1 (VCAM-1; CD106), platelet endothelial cell adhesion molecule-1 (PECAM-1; CD31), HLA-DR and E-selectin are expressed on endothelial cells, while intercellular adhesion molecule-1 (ICAM-1; CD54), very late antigen-4 (VLA-4), L-selectin and HLA-DR are expressed by inflammatory cells (see Ch. 102 ) .
Neutrophils are often numerous in early lesions, and it has been shown that a higher percentage of circulating neutrophils in patients with erythema nodosum leads to the production of reactive oxygen intermediates; these intermediates, in turn, may provoke inflammation and tissue damage . Support for a pathogenic role for these cells and molecules is provided by studies on the effects of colchicine . This inhibitor of neutrophil chemotaxis has been shown to diminish L-selectin expression on the neutrophil surface, inhibit E-selectin-mediated endothelial adhesiveness for neutrophils, and diminish stimulated expression of ICAM-1 on the endothelium .
Additional indirect evidence for the role of inflammatory cells and mediators includes reports of erythema nodosum following treatment with granulocyte colony-stimulating factor , and improvement (as well as flares) of erythema nodosum lesions with administration of tumor necrosis factor (TNF) inhibitors . Erythema nodosum, especially in its chronic phase, is characterized by granuloma formation and TNF is known to play a role in granuloma formation. A link between deregulation of TNF-α production and granuloma formation is further supported by the strong correlation of a polymorphism in the promoter region of the gene that encodes TNF-α and the development of sarcoidosis-associated erythema nodosum .
A wide range of precipitating factors has been linked with erythema nodosum. Infectious causes are common, particularly upper respiratory infections (both streptococcal and non-streptococcal). Other commonly reported causes are listed in Table 100.3 .
| CAUSES OF ERYTHEMA NODOSUM | ||
|---|---|---|
| Incidence | Cause | Comments |
| Most common | Idiopathic | Still the largest single category, accounting for a third to a half of cases |
| Streptococcal infections, especially of the upper respiratory tract | The largest single infectious cause | |
| Other infections: Viral upper respiratory tract infections Bacterial gastroenteritis – Yersinia > Salmonella, Campylobacter | Overall, infection may account for a third or more of cases | |
| Coccidioidomycosis | Erythema nodosum is associated with a lower incidence of disseminated disease | |
| Drugs * | Especially estrogens and oral contraceptive pills; also sulfonamides, penicillin, bromides, iodides; occasionally, TNF inhibitors, BRAF inhibitors ^ | |
| Sarcoidosis ** | 10–20% of cases in some series | |
| Inflammatory bowel disease | Crohn disease has a stronger association with erythema nodosum than does ulcerative colitis | |
| Uncommon | Infections: Brucella melitensis Chlamydophila † pneumonia Chlamydia trachomatis Mycoplasma pneumoniae Mycobacterium tuberculosis Histoplasma capsulatum Hepatitis B virus ‡ | |
| Neutrophilic dermatoses: Behçet disease Sweet syndrome | “Erythema nodosum” in Behçet disease more closely resembles erythema induratum (nodular vasculitis) | |
| Acne fulminans, including isotretinoin-associated | ||
| Pregnancy | ||
| Rare | Pernicious anemia | |
| Diverticulitis | ||
| Infections: Neisseria gonorrhoeae , N . meningitidis Escherichia coli , Bartonella henselae , Bordetella pertussis Treponema pallidum Dermatophytes (kerion), Blastomyces dermatitidis HIV Giardiasis, amoebiasis (abscesses) | Erythema nodosum leprosum is a different disease that is characterized by a cutaneous small vessel vasculitis | |
| MonoMAC syndrome ( GATA2 mutations) | ||
| Malignancy, most often acute myelogenous leukemia, Hodgkin disease | May overlap with Sweet syndrome | |
| Lupus erythematosus | One of several forms of panniculitis reported in LE, in addition to lupus panniculitis | |
* To be distinguished from panniculitis that can develop at sites of injection of medications, e.g. glatiramer acetate, interferon-β, phytonadione (vitamin K), interleukin-2, heparin (eosinophilic panniculitis), pentazocine, vaccines (e.g. tetanus).
** Löfgren syndrome is an acute, spontaneously resolving form of sarcoidosis characterized by erythema nodosum, hilar lymphadenopathy, fever, polyarthritis and uveitis.
^ Also lobular neutrophilic panniculitis.
† Previously referred to as Chlamydia .
‡ Erythema nodosum secondary to the hepatitis B vaccine has also been reported.
Clinical features
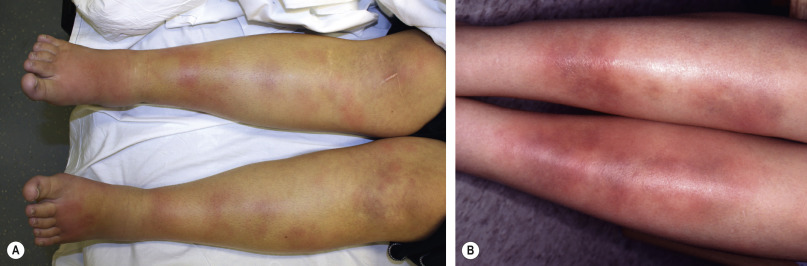
Erythema nodosum presents with bilateral, tender, erythematous nodules. These arise in crops and clearly the most common site is the shins ( Fig. 100.2 ). Other locations are occasionally involved, particularly the thighs and forearms . Nodules may also appear on the trunk, neck and face , but this is sufficiently rare that development of lesions in these locations should prompt consideration of other diagnoses. Unlike other forms of panniculitis, ulceration is not a feature of erythema nodosum. Systemic symptoms may occur that are not necessarily related to a specific coexisting systemic disorder; these include arthritis, arthralgia, fever and malaise .
Because of its close association with a variety of disorders and infections, erythema nodosum is an important skin sign of systemic disease. For example, its development may precede or accompany a flare of inflammatory bowel disease . It may also have some value as a prognostic indicator in certain disorders. For example, erythema nodosum is associated with a protective effect against disseminated disease in patients with coccidioidomycosis, and it is closely aligned with a more benign and self-limited form of sarcoidosis . Nevertheless, a significant percentage of cases – more than one-third – have no known disease association, even when followed for a year or more . Clinical or laboratory data that tend to predict that the development of erythema nodosum may be secondary to a systemic disease are listed in Table 100.4 .
| FINDINGS SUGGESTIVE OF A SYSTEMIC CAUSE FOR ERYTHEMA NODOSUM |
|---|
|
* Check for fecal white blood cells and stool culture for bacteria and, if indicated, ova and parasites.
Erythema nodosum lesions usually last a few days or weeks and then slowly involute, without scar formation. Discoloration suggestive of a bruise may be seen as the erythema subsides. More chronic forms do occur, some of which show a tendency toward migration or centrifugal spread; the latter have been termed subacute nodular migratory panniculitis or erythema nodosum migrans (see below). Up to one-third of cases of erythema nodosum recur. Annual recurrences are particularly common among idiopathic cases .
Pathology
Erythema nodosum is the prototypic septal panniculitis ( Fig. 100.3 ), but this should not be taken to imply that histopathologic changes are entirely confined to subcutaneous septa . Biopsy specimens of early lesions tend to show edematous septa and mild lymphocytic infiltrates. Of note, neutrophils may predominate in early lesions , and a variant with a predominance of eosinophils has been reported . True vasculitis of the type seen in leukocytoclastic vasculitis is not observed, and erythema nodosum is not generally regarded as a vasculitic process. However, “secondary” vasculitis may be observed in lesions when they contain relatively heavy, mixed, or neutrophil-rich inflammatory infiltrates. Erythema nodosum-like lesions in Behçet disease may demonstrate leukocytoclastic or lymphocytic vasculitis involving subcutaneous venules or muscular veins; the latter changes are prone to occur in patients with more severe forms of Behçet disease .
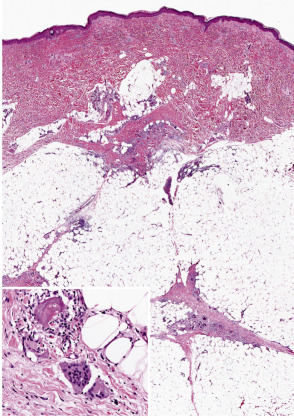
In early lesions, one may also find Miescher microgranulomas, a characteristic if not pathognomonic feature of erythema nodosum. These are small collections of macrophages, found within septa or at a septal–lobular interface, that tend to surround neutrophils or small cleft-like spaces . Reported variations in the frequency of these granulomas in erythema nodosum may result in part from differences in definition, in the acceptance of subtle changes, and in the rigor of the search. In older lesions, Miescher microgranulomas may feature epithelioid and multinucleated giant cells ( Fig. 100.3 , inset).
As lesions progress, the septa become widened and contain a mixed, partly granulomatous infiltrate. These cells infiltrate the periphery of fat lobules in a lace-like configuration. The extent of lobular involvement may vary, and in some cases can be prominent . Nevertheless, in the case of a lobular panniculitis without the characteristic septal changes, a diagnosis of erythema nodosum should be made with caution. Frequently, there is also a mild to moderate perivascular lymphocytic infiltrate in the overlying dermis. In later stages, the septa become fibrotic, partially replacing the fat lobules. Residual granulomas and lipophages can be observed, and a degree of vascular proliferation may be present . Over the long term, a remodeling process takes place that usually results in minimal residual scarring .
Differential diagnosis
The clinical scenario of an acute eruption of tender subcutaneous nodules over both shins of a young person is highly characteristic of erythema nodosum. However, when lesions are few in number, are located in sites other than the lower legs, or are of longer duration (>6 weeks), erythema nodosum can be difficult to distinguish from other forms of panniculitis. Lesions of erythema induratum (nodular vasculitis) can resemble those of erythema nodosum, but they tend to occur on the posterior aspect of the lower legs and may ulcerate. Ulceration is also a feature of pancreatic panniculitis, which occurs more frequently in other locations (although still favoring the lower legs), is more likely to be accompanied by arthritis and serositis, and is associated with elevated serum amylase and lipase levels.
Histopathologically, the picture of a predominantly septal panniculitis usually limits the differential diagnosis and tends to exclude those conditions that are chiefly lobular or mixed. Pancreatic panniculitis may show predominantly septal changes in its earliest stages , but eventually, these lesions exhibit the characteristic fat necrosis, with saponification and “ghost cell” formation. Infection-induced panniculitis can sometimes mimic erythema nodosum, but there are often more extensive neutrophilic infiltrates, cellular necrosis (including sweat gland necrosis), vascular proliferation, and hemorrhage .
Treatment
Treatments most often recommended for uncomplicated erythema nodosum include bed rest, salicylates, and nonsteroidal anti-inflammatory drugs (NSAIDs; Table 100.5 ) . Potassium iodide has been used with success, with adult dosages ranging from 450 to 1500 mg/day ( Table 100.6 ) . Improvement can be seen within 2 weeks. Potassium iodide may work through inhibition of cell-mediated immunity, as well as via inhibition of neutrophil chemotaxis and suppression of neutrophil-generated oxygen intermediates . In light of this treatment response, reports of erythema nodosum triggered by potassium iodide seem contradictory.
| USE OF POTASSIUM IODIDE (KI) |
|---|
| Saturated solution of potassium iodide (SSKI) |
|
| Side effects of SSKI |
|
* 0.3 ml = 10 drops from the calibrated dropper supplied with SSKI.
Treatment of erythema nodosum is influenced by underlying conditions. Thus, colchicine is useful in management of erythema nodosum that accompanies Behçet disease . Various treatments for inflammatory bowel disease are also effective in managing coexistent erythema nodosum . Both etanercept and infliximab have been reported to be effective in treating erythema nodosum , but paradoxically both have been noted to produce erythema nodosum as a cutaneous side effect . In case reports, adalimumab has led to improvement of refractory chronic erythema nodosum . Additional systemic therapies are listed in Table 100.5 .
Subacute Nodular Migratory Panniculitis
▪ Erythema nodosum migrans ▪ Chronic erythema nodosum
- ▪
Nodules on the lower extremities that migrate or undergo centrifugal spread, with central clearing
- ▪
Often unilateral
- ▪
Most cases are idiopathic; occasionally associated with streptococcal infection or thyroid disease
- ▪
More chronic course than typical erythema nodosum
Clinical features
This condition was first described by Bafverstedt in 1954 and was named subacute nodular migratory panniculitis by Vilanova and Piñol Aguade in 1956 . Some of its clinical and microscopic characteristics are similar to chronic erythema nodosum, and it is believed by many to represent a variant of the latter . However, others consider it to be a separate disorder . Subacute nodular migratory panniculitis is seen predominantly in women, is often unilateral, and is characterized by nodules that migrate or expand in a centrifugal manner (with central clearing) and may assume a yellowish or morpheaform appearance . Lesions tend to be less tender than those of classic erythema nodosum. There may be few, if any, associated systemic symptoms , though arthralgias have been reported and the ESR may be elevated . Most cases are idiopathic, but some are associated with streptococcal infection (as evidenced by elevated antistreptolysin O and anti-DNase B titers) or thyroid disease .
Pathology
Microscopically, the changes are those of a chronic septal panniculitis . However, in contrast to more classic forms of chronic erythema nodosum, subacute nodular migratory panniculitis shows greater septal thickening, more prominent granulomatous inflammation along the borders of widened subcutaneous septa, absence of phlebitis, and rare hemorrhage .
Treatment
Untreated, subacute nodular migratory panniculitis can last for months or years. However, treatment with potassium iodide is usually effective, resulting in clearing of lesions within several weeks .
Morphea/Scleroderma Panniculitis
(See also Chs 43 & 44.)
Clinical features
Both morphea and scleroderma (systemic sclerosis) can affect the subcutaneous fat ( Table 100.7 ), which is the primary site of involvement in deep morphea (morphea profunda) . Subcutaneous extension also occurs in variants such as disabling pansclerotic morphea of childhood, generalized morphea, and linear morphea. In addition, eosinophilic fasciitis can involve the subcutaneous fat as well as the fascia . These syndromes may be associated with peripheral eosinophilia, polyclonal gammopathy, and serologic abnormalities . Fleischmajer et al. proposed that the sclerosing process in scleroderma is initiated by the changes that take place in the subcutis.
| CLINICAL AND MICROSCOPIC FEATURES OF CONNECTIVE TISSUE PANNICULITIS | ||||
|---|---|---|---|---|
| Disorder | Clinical presentation | Lesion distribution | Relation to systemic disease | Histopathology of panniculitis |
| Morphea and scleroderma (systemic sclerosis) | Indurated plaques | Extremities, trunk | Occurs in scleroderma, but usually more pronounced in morphea | Septal, with thickening and sometimes mucin deposition; inflammation especially at dermal–subcutaneous interface; lymphocytes and plasma cells; lymphoid follicles in morphea * |
| Lupus panniculitis | Tender subcutaneous nodules and plaques; may have overlying lesions of discoid LE | Face (especially cheeks), upper arms, shoulders, hips, buttocks, breasts, trunk | Only minority of patients have associated systemic LE | Lobular or mixed lobular/septal; mucin deposition; hyaline necrosis of lobules; lymphocytes and plasma cells, sometimes with prominent nuclear dust; nodular lymphocytic aggregates or lymphoid follicles |
| Dermatomyositis | Indurated, painful plaques and nodules; may ulcerate | Buttocks, abdomen, thighs, arms | Can arise in patients with established dermatomyositis or precede other disease manifestations | Lobular or mixed septal/lobular; fat necrosis; lipomembranous changes; sometimes calcification; lymphocytes and plasma cells; sometimes nodular lymphocytic aggregates |
| Annular atrophic connective tissue panniculitis of the ankles | Subcutaneous atrophy, which may be preceded by induration and tenderness | Circumferential bands around the ankles | Patients often have antinuclear antibodies and/or autoimmune conditions such as thyroiditis or rheumatoid arthritis | Lobular or mixed septal/lobular lymphohistiocytic panniculitis with areas of fat necrosis |
| Atrophic connective tissue panniculitis | Erythematous, indurated plaques that resolve with subcutaneous atrophy | Favors extremities; may be widespread | As above | As above |
Pathology
Table 100.7 outlines the microscopic changes in morphea- and scleroderma-associated septal panniculitis ( Fig. 100.4 ). Lymphocytes and plasma cells predominate , although macrophages and eosinophils may be present. In some cases, the number of plasma cells is striking . In late stages of morphea, the subcutis is largely replaced by hyalinized connective tissue, very often accompanied by changes of lipoatrophy.
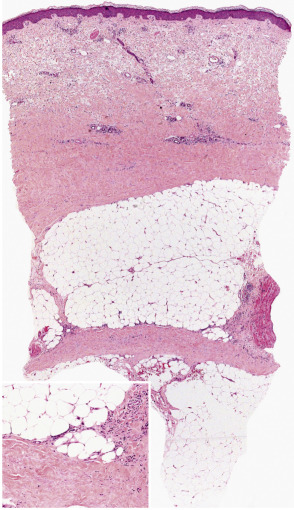
Differential diagnosis
The combination of septal panniculitis with lymphoplasmacytic predominance and dermal and subcutaneous sclerosis is unique, and this helps to separate morphea/scleroderma panniculitis from other septal forms of panniculitis. Differentiation among the several variants outlined above is more difficult, requiring clinical data for accurate classification. Sclerosis with lesser degrees of subcutaneous inflammation characterizes scleroderma, while a predominance of fascial involvement with extension into the subcutis is more typical of eosinophilic fasciitis.
Alpha-1 Antitrypsin Deficiency Panniculitis
▪ Alpha-1 protease (proteinase) deficiency panniculitis
- ▪
Erythematous, painful, subcutaneous nodules or plaques that often ulcerate and drain
- ▪
Associated with alpha-1 antitrypsin deficiency; patients with the most severe disease are homozygotes for the Z allele of the SERPINA1 gene (PiZZ)
- ▪
A characteristic histologic finding is liquefactive necrosis of the dermis and subcutaneous septa, but lobular or mixed septal–lobular changes with neutrophils may occur
Introduction
Alpha-1 antitrypsin deficiency is a well-established, but uncommon, cause of panniculitis. The most severely affected individuals, with markedly decreased levels of the protease inhibitor, are most prone to the development of ulcerating neutrophilic panniculitis. Recognition of the disorder is important not only in the selection of appropriate therapy, but also in addressing other systemic manifestations of the disease and in dealing with its genetic aspects.
History
Alpha-1 antitrypsin deficiency is an inborn error of metabolism that was first delineated by Eriksson and others in the early 1960s . In 1972, Warter and colleagues identified members of a family with alpha-1 antitrypsin deficiency and “Weber–Christian syndrome”. Subsequent investigations linked the clinical and microscopic findings to known effects of proteinase inhibitor deficiency.
Epidemiology
No apparent racial or geographic prevalence has been noted for alpha-1 antitrypsin deficiency panniculitis. The incidence of the disease is approximately equal in men and women . Age of onset ranges from infancy to the eighth decade of life .
Pathogenesis
Alpha-1 antitrypsin, a glycoprotein produced in the liver, is the most abundant circulating ser ine p rotease in hibitor (serpin). The more than 120 different alleles of the gene that encodes this protein (SERPINA1; formerly known as PI) are divided into categories based upon the electrophoretic mobility of their protein products (M = medium, S = slow, Z = very slow). The most common protease inhibitor (Pi) phenotype is MM (homozygous for M alleles), which is associated with normal serum levels of alpha-1 antitrypsin (100–200 mg/dl) . Heterozygotes with one copy of the S or Z allele have mild to moderate deficiencies of the inhibitor (PiMS and PiMZ; prevalences of 1–3% in Caucasian populations). Patients who are homozygous for the Z allele (PiZZ; prevalence of 1 : 150–1 : 5000 in Caucasian populations) or whose genotype is the rare PiZnull have severe alpha-1 antitrypsin deficiency, with serum levels in the range of 20–45 mg/dl. In these individuals, most of the aberrant alpha-1 antitrypsin protein accumulates in the endoplasmic reticulum of hepatocytes, and the small amounts that enter the circulation have decreased function and a tendency to form inactive polymers that may stimulate neutrophil chemotaxis . Of note, Z-type polymers have been detected in the skin of a patient with alpha-1 antitrypsin deficiency panniculitis, further suggesting a possible proinflammatory role .
Alpha-1 antitrypsin acts upon a wide range of proteolytic enzymes that play a direct role in degradation of tissues, including trypsin, collagenase, and elastase . It also has important effects on immune function, e.g. inhibition of membrane-bound serine proteases involved in the activation of lymphocytes and macrophages . It may also inhibit complement activation, both through a direct effect on complement-related proteases and by inhibiting the neutrophil proteases that activate enzymes of the complement system .
In addition to panniculitis, the consequences of alpha-1 antitrypsin deficiency include chronic liver disease with cirrhosis (resulting from retention of the aberrant protein within the liver), emphysema, pancreatitis, membranoproliferative glomerulonephritis, rheumatoid arthritis, c-ANCA (cytoplasmic antineutrophil cytoplasmic antibody)-positive vasculitis, other cutaneous vasculitides such as Henoch-Schönlein purpura, and angioedema (resulting from deficiency of the protease inhibitor) . The initiating event in individuals who develop panniculitis is not always clear; trauma appears to play a role in some patients. Postpartum flares of the disease have been reported in genetically susceptible individuals. This is attributed to the estrogen-promoted increase in proteinase inhibitor levels during pregnancy, followed by a precipitous decline to subnormal levels postpartum .
Absence of the alpha-1 antitrypsin protease inhibitor results in activation of lymphocytes and macrophages, lack of restraint upon the complement cascade, release of chemotactic factors, accumulation of neutrophils with release of their proteolytic enzymes, and consequent attack upon fat and nearby connective tissues . The subcutis may be particularly vulnerable to this process, since fatty acids make nearby elastin more susceptible to proteolytic degradation .
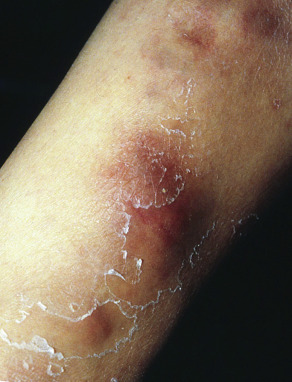
Clinical features
Large, erythematous to purpuric, tender nodules or plaques appear in a variety of sites ( Fig. 100.5 ), especially the lower trunk and proximal extremities (flanks, buttocks and thighs) . Ulcers develop that may be deep and necrotic, accompanied by an oily discharge . Migration of lesions has been reported . A history of antecedent trauma can be elicited in approximately one-third of patients . Panniculitis may be accompanied by fever, pleural effusions and pulmonary emboli . The clinical course of the panniculitis is often prolonged, and lesions are resistant to immunosuppressive therapy. Healing is accompanied by scarring and subcutaneous atrophy . The most severe manifestations arise in those with profound proteinase inhibitor deficiency (PiZZ), although the panniculitis can also occur in heterozygotes .
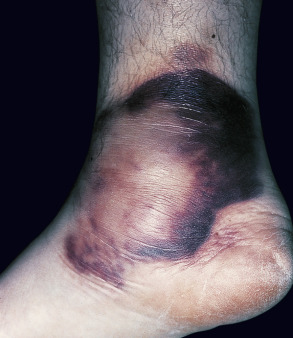
Pathology
Descriptions of the pathology of alpha-1 antitrypsin deficiency panniculitis have varied. Early, there is a neutrophilic panniculitis, followed rapidly by necrosis and destruction of fat lobules . Splaying of neutrophils between collagen bundles in the reticular dermis has been described as an early clue to the diagnosis . Dissolution of dermal collagen, with resultant liquefactive necrosis and separation of fat lobules from adjacent septa, is a principal change in most cases . Another characteristic feature is the presence of “skip areas” of normal fat adjacent to foci of severe necrotizing panniculitis . Chronic inflammation and hemorrhage may be present at the periphery of areas of involvement . Most authors have not found evidence for primary leukocytoclastic vasculitis, although there may be evidence for lymphocytic vasculitis, secondary vasculitis in areas of heavy neutrophilic infiltration, or thrombosis . In individuals with intermediate levels of protease inhibitor deficiency, lipophage and giant cell accumulation may be prominent . Lesions heal with scarring and obliteration of fat lobules.
Views differ on whether alpha-1 antitrypsin deficiency panniculitis should be regarded as a primarily septal or lobular panniculitis. Clearly, involvement of fat lobules can be significant, and, as a result, there are authors who have labeled the process a lobular panniculitis. On the other hand, some descriptions have emphasized early septal inflammation, collagenolysis of the fibrous septa ( Fig. 100.6 ) , and the prominent septal fibrosis in late-stage lesions .
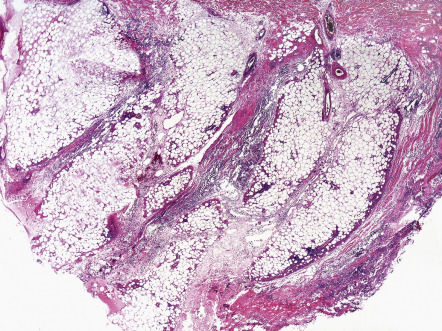
Differential diagnosis
Clinically, the degrees of inflammation, ulceration and drainage associated with alpha-1 antitrypsin deficiency panniculitis may actually elicit a differential diagnosis more focused upon ulcerative skin disorders (see Fig. 105.1 ). Ulcers associated with alpha-1 antitrypsin deficiency generally lack the necrotic, “undermined” borders associated with pyoderma gangrenosum . Tissue culture to exclude infection-induced panniculitis may be required.
Entities in the microscopic differential diagnosis include traumatic (factitial) panniculitis, infection-induced panniculitis, pancreatic panniculitis, and erythema induratum (nodular vasculitis) . Each of these can be associated with infiltrates that include neutrophils and varying degrees of necrosis, yet each has other distinct findings (see below). In the appropriate clinical setting, subcutaneous Sweet syndrome and neutrophilic panniculitis in patients with rheumatoid arthritis or inflammatory bowel disease may represent additional diagnostic considerations.
Treatment
Treatments that are usually ineffective include corticosteroids and other immunosuppressants, cytotoxic agents, colchicine, danazol, and hydroxychloroquine . Doxycycline is sometimes effective, particularly in mild cases; one suggested dosage schedule is 200 mg twice daily for 3 months . Dapsone may also be beneficial in mild cases, by suppressing neutrophil migration and inhibiting oxidation reactions induced by myeloperoxidase . Reduction of alcohol intake has also been recommended, since ethanol (as a hepatotoxin) may precipitate alpha-1 antitrypsin-associated hepatitis .
The most effective therapeutic intervention is replacement of alpha-1 antitrypsin via intravenous infusions. Dosages are generally 60 mg/kg per week, administered over a period of 3 to 7 weeks . Improvement is relatively rapid, in that clearing of the panniculitis can occur after three weekly doses . Recurrences are possible when alpha-1 antitrypsin levels fall below 50 mg/dl , but these typically respond to further replacement therapy. Other successful therapies include plasma exchange and liver transplantation (in appropriate clinical circumstances). There is evidence that autophagy-enhancing drugs, such as carbamazepine, may reduce systemic manifestations, including hepatic fibrosis , and clinical trials are currently underway .
Erythema Induratum
▪ Nodular vasculitis ▪ Erythema induratum ofBazin or Bazin disease (tuberculous etiology) ▪ Erythema induratum of Whitfield (non-tuberculous etiology)
- ▪
Erythematous nodules or plaques, usually on the posterior lower legs of young to middle-aged women
- ▪
Ulceration and drainage may occur
- ▪
Microscopic features of lobular or mixed panniculitis with evidence of vasculitis involving arteries or veins
- ▪
Classically associated with tuberculosis, but similar lesions can be idiopathic or induced by other infectious agents or drugs
Introduction
Erythema induratum is a condition characterized by nodules on the lower extremities, which may ulcerate and drain. Originally regarded as a tuberculid, its relationship to tuberculosis in a subset of patients has been solidified by more recent studies that have detected mycobacterial DNA in cutaneous lesions.
History
Erythema induratum was first described by Ernest Bazin in 1861. It was generally regarded as a tuberculid because of a strong association with tuberculosis, although Koch’s postulates could not be fulfilled. In 1945, Montgomery and colleagues proposed the term nodular vasculitis for cases with similar clinical and pathologic features that were not of tuberculous origin. Since the early 1970s, erythema induratum and nodular vasculitis have generally been considered as synonyms referring to a clinicopathologic entity with several possible causes, one of which is tuberculosis. The detection of mycobacterial DNA in cutaneous lesions has confirmed a tuberculous origin in some patients . There are certain clinicians, however, who prefer to use the term nodular vasculitis when referring to individuals with a non-tuberculous etiology.
Epidemiology
In erythema induratum, an overwhelming female predominance is observed, but men can also develop the disease . There is no apparent racial predilection, and although there is a wide age range among affected patients, the mean is between 30 and 40 years . Erythema induratum of tuberculous etiology occurs more frequently in populations with a high prevalence of tuberculosis.
Pathogenesis
As mentioned previously, some cases have been strongly associated with Mycobacterium tuberculosis infection. This can be substantiated by the detection of mycobacterial DNA in skin lesions by PCR , with specific primers used to distinguish M. tuberculosis complex DNA from that of other mycobacterial pathogens . Non-tuberculous cases have been related to other infectious agents (e.g. Nocardia , hepatitis C virus ) or to drugs (e.g. propylthiouracil ). There is a report of erythema induratum being induced by a tuberculin skin test.
Although it has been suggested that erythema induratum results from an immune complex-mediated vasculitis , most investigators believe that the process represents a type IV, cell-mediated response to an antigenic stimulus . Biopsy specimens show a predominance of T lymphocytes, macrophages and dendritic cells, including Langerhans cells . One study of peripheral blood mononuclear cells from a patient with erythema induratum and active tuberculosis showed a high proliferative response to purified protein derivative (PPD) and marked production of interferon-γ, suggesting a pathogenic role for these PPD-specific T cells in a delayed hypersensitivity response to mycobacterial antigens .
Clinical features
Erythema induratum is characterized by tender, erythematous to violaceous nodules and plaques that most often develop on the lower legs, especially the calves . Lesions have also been reported on the feet, thighs, buttocks and arms . An annular arrangement of nodules has been described in M. tuberculosis -related cases . Ulceration can occur ( Fig. 100.7 ). Lesions are persistent, tend to heal with scarring, and are prone to recurrence . In erythema induratum associated with M. tuberculosis , there may be clinical and radiographic evidence for active tuberculosis, positive skin tests to PPD, or a positive interferon-gamma release assay such as the QuantiFERON ® -TB Gold In-Tube test . In addition, other tuberculids, e.g. papulonecrotic, may be present. Clinical differences between tuberculous and non-tuberculous cases are minor.
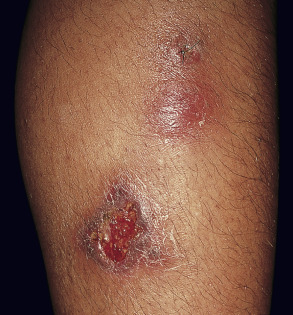
Pathology
Erythema induratum is generally described as a lobular or mixed septal/lobular panniculitis. Inflammation is mixed, and can include neutrophils, lymphocytes, macrophages and multinucleated giant cells ( Fig. 100.8A ). Vasculitis is identifiable in the vast majority of cases, and most frequently involves veins or arteries of connective tissue septa and small venules of the fat lobules ( Fig. 100.8B ). It may be predominantly neutrophilic , lymphocytic , or granulomatous. Necrosis with a coagulative or caseous appearance may be present, sometimes with palisading granulomas . Necrosis has been described in both tuberculous and non-tuberculous cases, and the incidence and degree of necrosis are greater in those cases that are positive for M. tuberculosis DNA by PCR methods . However, this finding is absent in over half of cases.
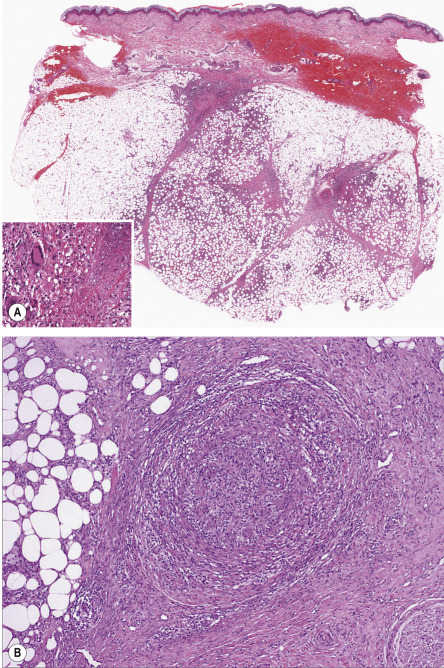
Differential diagnosis
Infection-induced panniculitis tends to show a more prominent neutrophilic component, granular basophilic necrosis, sweat gland necrosis and proliferation of small vessels, and organisms may be identified on special staining. Lupus panniculitis tends to be less granulomatous, has a prominent lymphoplasmacellular infiltrate, may show mucin deposits, and sometimes has overlying epidermal and dermal changes typical for lupus erythematosus. Both polyarteritis nodosa and thrombophlebitis tend to show inflammation limited to the immediate perivascular zone, in contrast to the extensive lobular panniculitis often encountered in erythema induratum. Histologically, perniosis can be difficult to distinguish from erythema induratum, but there is typically a history of cold exposure , and on microscopic examination prominent involvement of dermal vessels is often observed, with “fluffy edema” of their walls.
Treatment
Treatment should be directed at the underlying cause, if found. This includes multi-drug antituberculous therapy (see Ch. 75 ) and discontinuing possible inciting medications. Therapeutic options for nontuberculous erythema induratum include NSAIDs, corticosteroids, tetracyclines, potassium iodide, and mycophenolate mofetil . Supportive care is similar to that for erythema nodosum (see Table 100.5 ).
Pancreatic Panniculitis
▪ Pancreatic fat necrosis ▪ Enzymatic panniculitis
- ▪
Subcutaneous nodules, sometimes accompanied by fever, arthritis or abdominal pain
- ▪
Associated with pancreatic disorders, including acute and chronic pancreatitis and pancreatic carcinoma
- ▪
Mixed septal/lobular panniculitis featuring “ghost cell” formation and the deposition of basophilic material due to saponification of fat by calcium salts
- ▪
Treatment primarily directed towards the underlying pancreatic disorder
Introduction
Pancreatic panniculitis is an unusual complication of pancreatic disease. In addition to the symptoms associated with fat necrosis, its chief importance is as a sign of a significant systemic disorder, particularly because the panniculitis may be recognized prior to detection of the underlying pancreatic disease.
History
Chiari first described pancreatic panniculitis in 1883. By 1999, fewer than 100 cases had been reported . Since then, an additional 90 publications have appeared, primarily as case reports.
Epidemiology
Panniculitis develops in up to 2% of patients with pancreatic disorders . No geographic, racial or gender predilections have been reported.
Pathogenesis
There is considerable evidence that the enzymes lipase, amylase and trypsin are involved in producing the lesions of pancreatic panniculitis . Elevated enzyme levels have been detected in the blood , urine , and skin lesions, even in the absence of detectable pancreatic disease. Lipase has the clearest relationship with the panniculitis, with a number of patients having elevated serum lipase levels but normal amylase levels . Utilizing an anti-pancreatic lipase monoclonal antibody, positive intracellular adipocyte staining was observed in a biopsy of lesional skin .
Amylase levels, when elevated, tend to peak 2–3 days after eruption of the skin lesions and return to normal 2–3 days after regression of the lesions. Trypsin and perhaps amylase may act by promoting increased permeability of vessel walls, thereby permitting lipase to hydrolyze neutral fat to form glycerol and free fatty acids, with resulting fat necrosis and inflammation . Venous stasis may promote this process, possibly explaining the predilection for the lower extremities. Elevated enzyme levels may not be the complete explanation for the changes of pancreatic panniculitis ; immunologic factors probably also play a role.
Clinical features
Subcutaneous nodules develop in association with acute or chronic pancreatitis, pancreatic carcinoma (acinar cell > other types [e.g. neuroendocrine carcinomas]), pancreatic pseudocysts, pancreas divisum, or traumatic pancreatitis . The possibility of a pancreaticoportal fistula should be considered when panniculitis develops in individuals with chronic pancreatitis, and in some of these patients, an acinar cell carcinoma can also be found. Panniculitis may precede detection of pancreatic disease by 1–7 months , and in the case of pancreatic carcinoma, its onset may signal the presence of metastatic disease .
In pancreatic panniculitis, subcutaneous nodules develop, frequently on the legs but also on the anterior trunk, arms ( Fig. 100.9 ), and scalp . Erythematous, edematous, sometimes painful lesions arise singly or in crops, and they may migrate. They can become fluctuant and ulcerate, discharging an oily material . Panniculitis may also involve visceral fat, including the omentum and preperitoneal fat . Associated findings include fever, abdominal pain, inflammatory polyarthritis, ascites and pleural effusions .