Abstract
Porphyrias are a group of inherited or acquired metabolic disorders manifested by cutaneous photosensitivity, episodic neurovisceral dysfunctions, or both. Each porphyria is caused by reduced activity (or in one case, increased function) of one of eight enzymes regulating the porphyrin–heme pathway. These enzyme anomalies produce accumulations of excess porphyrin precursors and/or porphyrins in patterns characterizing each associated porphyria. Many mutations in the genes encoding the protein structures of these enzymes have been identified, with various aberrations of each gene leading to clinical expression of its associated porphyria. The severity of the encoded protein defect typically determines how much, if any, residual enzyme activity remains. Degrees of phenotypic severity may be linked to variations in molecular genotype. Clinical expression of some porphyrias may also be influenced by environmental and physiologic factors. Most porphyrias are readily diagnosed biochemically. Mutation analysis of the genes underlying any of the heritable porphyrias can be obtained commercially. Only porphyrias with cutaneous manifestations are reviewed in this chapter.
Keywords
Congenital erythropoietic porphyria, Erythropoietic protoporphyria, Hepatoerythropoietic porphyria, Hereditary coproporphyria, Porphyria cutanea tarda, Variegate porphyria, X-linked dominant protoporphyria
- •
Several porphyrias share similar photocutaneous features; sufficient testing to assure correct diagnosis must precede selection of therapies.
- •
Most porphyrias can be correctly diagnosed biochemically.
- •
Mutation analysis is the gold standard for porphyria diagnosis and family counseling.
- •
Clinical and biochemical remissions can be therapeutically induced in most patients with porphyria cutanea tarda.
- •
X-linked dominant protoporphyria, recently recognized, can be distinguished from erythropoietic protoporphyria despite many similar clinical features.
- •
Attacks of both variegate porphyria and hereditary coproporphyria can be rapidly diagnosed by screening for increased urinary porphobilinogen.
- •
Management of porphyrias with multisystem problems may be complex, requiring expert consultation.
Porphyrias are a group of inherited or acquired metabolic disorders manifested by cutaneous photosensitivity, episodic neurovisceral dysfunctions, or both ( Table 28-1 ). Each porphyria is caused by reduced activity (or in one case, increased function) of one of eight enzymes regulating the porphyrin–heme pathway ( Fig. 28-1 ). These enzyme anomalies produce accumulations of excess porphyrin precursors and/or porphyrins in patterns characterizing each associated porphyria.
| Cutaneous | Neurovisceral | Cutaneous and Neurovisceral | |
|---|---|---|---|
| Hepatic | Porphyria cutanea tarda Hepatoerythropoietic porphyria | Acute intermittent porphyria δ-Aminolevulinic acid dehydrogenase deficiency porphyria | Hereditary coproporphyria Variegate porphyria |
| Erythropoietic | Erythropoietic protoporphyria X-linked dominant protoporphyria Congenital erythropoietic porphyria |
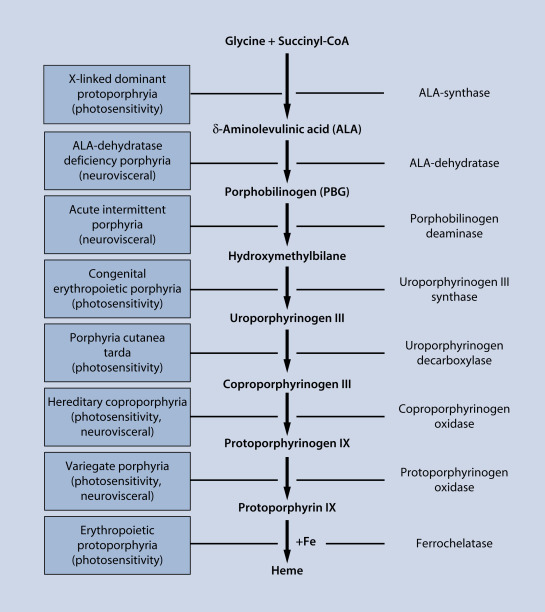
Human heme biosynthesis occurs chiefly in hepatocytes and bone marrow erythrocyte precursors. Porphyrias are classified as “hepatic” or “erythropoietic” according to the major tissue overproducing porphyrins and/or porphyrin precursors. Reduced enzyme activity causes backup accumulation of its immediate substrate and other precursors. Irreversible spontaneous oxidation of nonphotoactive porphyrinogen precursors forms photoactive porphyrin byproducts of the pathway that can be found in urine, feces, plasma and/or erythrocytes ( Table 28-2 ), and are disseminated throughout the body. Porphyrin molecules avidly absorb light energy of several wavelengths, maximally at 400 to 410 nm (visible violet light). Such energy can penetrate human epidermis and photoexcite porphyrins in dermal tissues. This excitation energy can then transfer to oxygen, yielding unstable, highly reactive oxidizing agents in the skin. Signs and symptoms of cutaneous photosensitivity result when lipid peroxidation and complement activation ensue, causing cell membrane disintegration and release of intravascular and cellular contents including secondary mediators of inflammation.
| Urine | Feces | Erythrocytes | Plasma | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Type of Porphyria | ALA | PBG | URO | COPRO | URO | COPRO | PROTO | URO | COPRO | PROTO | |
| Acute Porphyrias | |||||||||||
| Acute intermittent porphyria | ++ to ++++ | ++ to ++++ | +++ | ++ | N to + | N to + | N to + | N | N | N | N |
| Variegate porphyria | ++ to +++ | ++ to +++ | +++ | +++ | N | +++ | ++++ | N | N | N | 625–627 nm ∗ |
| Hereditary coproporphyria | N to ++ | N to ++ | ++ | ++++ | ++ | ++++ | N to + | N | N | N | 619 nm ∗ |
| ALA-D deficiency porphyria | +++ | N | + | ++ | N | + | + | N | N | ++ | ALA, COPRO and PROTO↑ |
| Nonacute Porphyrias | |||||||||||
| Porphyria cutanea tarda | N | N | ++++ | ++ | ++ | ISOCOPRO | + | N | N | N | URO↑ |
| Erythropoietic protoporphyria | N | N | N | N | N | ++ | ++ to ++++ | N | N to + | +++ | PROTO↑ |
| Congenital erythropoietic porphyria | N | N | ++++ | ++ | + | +++ | + | ++++ | +++ | +++ | URO and COPRO↑ |
| Hepatoerythro-poietic porphyria | N | N | +++ | ISOCOPRO | N | ISOCOPRO | N | N | + | ++++ | URO↑ |
| X-linked dominant protoporphyria | N | N | N | N | NA | NA | NA | NA | NA | ++++ † | PROTO↑ |
Four hepatic porphyrias manifest potentially fatal episodic attacks reflecting dysfunctions of central, autonomic, and peripheral nervous systems. Symptoms may include abdominal pain, limb pain and weakness, nausea, vomiting, constipation, tachycardia, hypertension, anxiety, agitation, confusion, bizarre behavior, central nerve paralysis, respiratory distress, seizures, or coma, which may be related to poorly understood effects of deranged heme synthesis on neurons. Attacks are often triggered by porphyrinogenic drugs that induce hepatic heme synthesis. Other precipitating factors include: fasting, infections, hormonal fluctuations due to menses, pregnancy or therapeutic hormones, or other stressors. Two acute attack porphyrias may also have photocutaneous features.
Many mutations in the genes encoding the protein structures of these enzymes have been identified, with various aberrations of each gene leading to clinical expression of its associated porphyria. Severity of the encoded protein defect typically determines how much, if any, residual enzyme activity remains. Degrees of phenotypic severity may be linked to variations in molecular genotype. Clinical expression of some porphyrias ( Table 28-1 ) may also be influenced by environmental and physiologic factors.
Most porphyrias are readily diagnosed biochemically ( Table 28-2 ). Enzyme activity assays are commercially available for only some enzymes of heme synthesis, but mutation analysis of the genes underlying any of the heritable porphyrias can be obtained. Several laboratories offering various analyses are listed at the American Porphyria Foundation website ( www.porphyriafoundation.com ). Only porphyrias with cutaneous manifestations will be further reviewed in this chapter.
Porphyria Cutanea Tarda
In porphyria cutanea tarda (PCT), the most common porphyria of adults, excess polycarboxylated porphyrins are produced due to reduced activity of uroporphyrinogen decarboxylase (UROD) in hepatocytes. PCT is the only porphyria with both acquired (∼80%, “sporadic” or “type 1”) and familial (∼20%, “type 2”) forms. Autosomal dominant familial PCT has a genotype of one mutated and one normal UROD allele. The resulting residual enzyme activity can be as low as 50% when the enzyme product of the mutant allele retains no activity whatsoever. Homozygous or compound heterozygous inheritance of two UROD mutations reduces residual activity to only 10% to 30% of normal, causing the more severe recessive variant “hepatoerythropoietic porphyria” (HEP). Many deleterious UROD mutations are known; the HEP genotype is often heteroallelic, with a different mutated allele inherited from each parent. While PCT usually presents in adulthood, HEP presents in childhood. The destructive photomutilation observed in HEP simulates that of congenital erythropoietic porphyria (CEP).
UROD sequentially decarboxylates uroporphyrinogen (eight carboxylic side groups) to coproporphyrinogen (four carboxylic side groups). Sufficiently reduced UROD activity leads to abnormally high levels of uroporphyrin and 7-carboxyl porphyrin, with lesser increases in 6- and 5-carboxyl porphyrins and coproporphyrin in urine and plasma, and abnormal fecal excretion of isocoproporphyrin, an unusual 4-carboxyl pathway by-product. In most familial PCT cases, UROD deficiency is detectable in both erythrocytes and hepatocytes; in acquired PCT, only hepatocytes have diminished UROD activity.
Clinical Manifestations
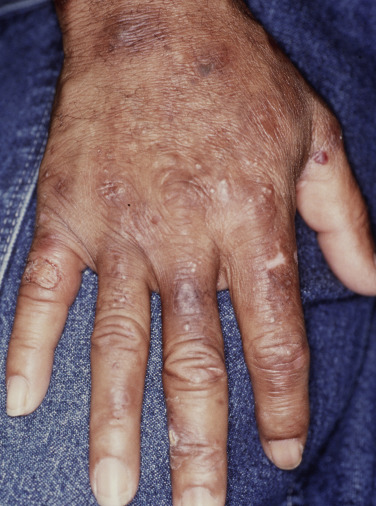
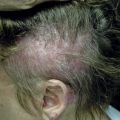
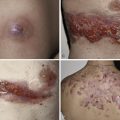
Cutaneous features of PCT predominantly involve sun-exposed skin of the dorsal hands, extensor forearms, and face ( Figs 28-2 to 28-5 ), but legs, feet, scalp, and chest ( Fig. 28-6 ) may also be affected. Vesicles, bullae ( Figs 28-2 and 28-3 ) and painful erosions of fragile skin ( Fig. 28-4 ) are typical presenting complaints. Facial hypertrichosis ( Fig. 28-5 ) frequently develops; mottled melasma-like dyspigmentation may appear in the periorbital and malar regions. Rarely, generalized melanosis simulates Addison’s disease or hemochromatosis. Sclerodermoid induration of head, neck, back, and chest skin, histopathologically indistinguishable from scleroderma, may be an accompanying or sole finding ( Fig. 28-7 ). Protracted blistering eventuates in scars, dyschromia, and milia ( Figs 28-2 and 28-8 ), as well as alopecia with distinctive saucer-shaped scars or crater-like depressions. Indolent ulcers and dystrophic calcification may develop in chronically damaged preauricular, forehead, scalp, neck, or dorsal hand skin.