Key points
- •
Both itch and pain are very frequent symptoms in patients with atopic dermatitis, with a prevalence of more than 50%.
- •
Patients with atopic dermatitis show neuronal sensitization to both itch and pain.
- •
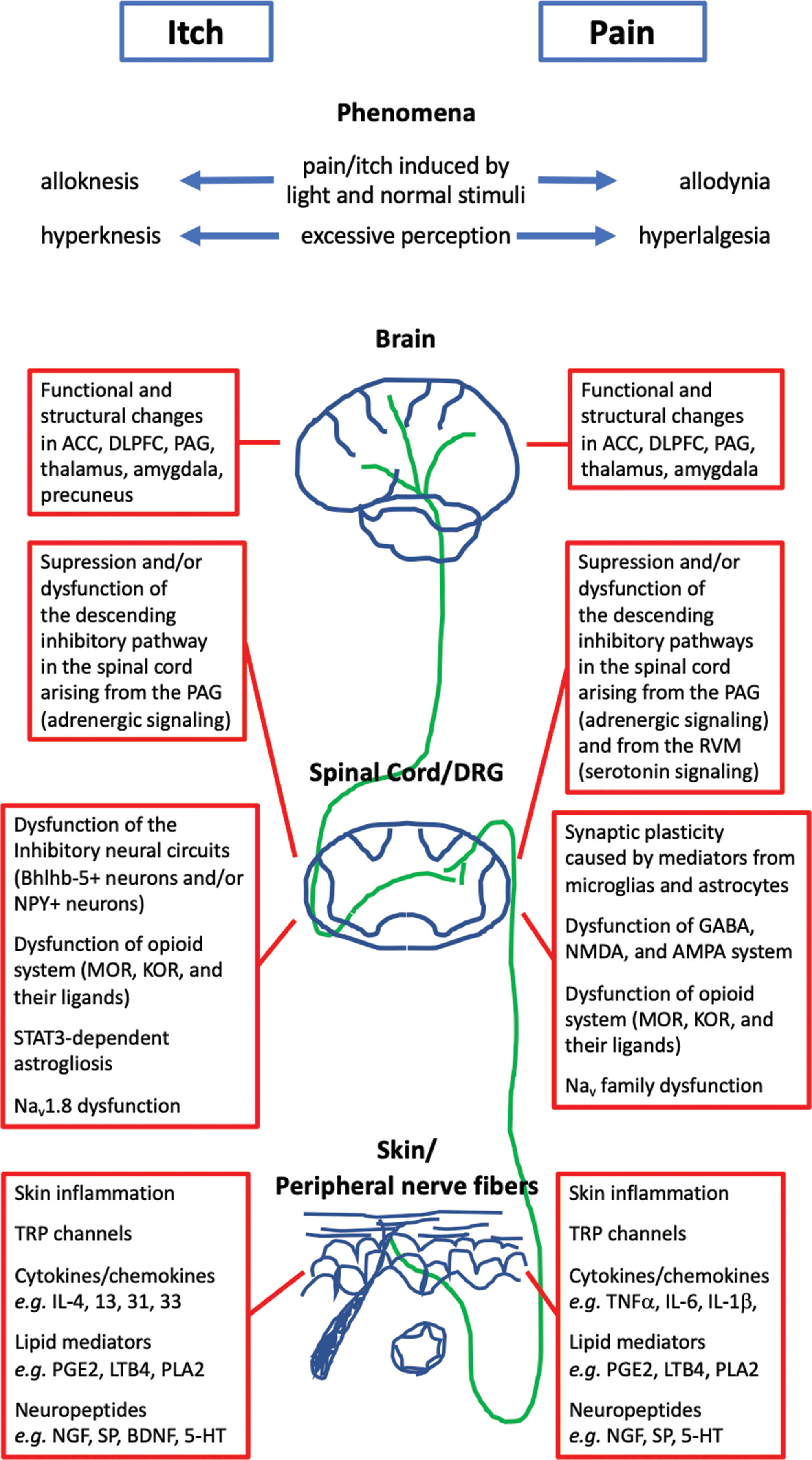
Itch neuronal sensitization is associated with alloknesis (phenomena in which normal or nonpruritic stimuli are perceived as itchy) and hyperknesis (excessive itch perception to pruritic stimuli).
- •
Pain neuronal sensitization is associated with hyperalgesia (increased pain due to a normal noxious stimuli) and allodynia (a painful response to normally nonnoxious mechanical, tactile, or thermal stimuli).
- •
Neuronal sensitization to itch and pain can occur at any level of perception, including peripheral sensitization (in the skin and peripheral sensory neurons) and central sensitization (at neuronal circuits in the spinal cord, descending inhibitory pathways in the spinal cord, and the brain).
Introduction
Itch and pain are two main symptoms that play major roles in causing morbidities in patients affected by atopic dermatitis (AD). In addition, the persistence of these symptoms can significantly affect the general health of afflicted patients and even patients’ family members. In this chapter we discuss the prevalence of these symptoms, the pathomechanisms of these symptoms, and the relationship between itch and pain as it occurs in AD.
Prevalence of itch and pain in atopic dermatitis
Itch is the predominant feature of AD. A study of 304 patients with AD revealed that itch occurred at least once a day in over 90% of patients with mean itch intensity of 8.3 out of 10 ( ). In another study of 170 members of the French Eczema Association, the worst pruritus intensity was 8.8 out of 10 ( ). Patient-reported outcomes collected at screening from a multinational, phase 2 clinical trial of dupilumab showed that 85% of patients reported itch with mean intensity of 6.5 out of 10 ( ).
Similar to itch, pain is one of the most frequent symptoms of AD. One study of 284 AD participants found that pain was the third most common symptom (58%) after itch (99%) and sleep difficulties (66%). Other studies also revealed a similar prevalence of pain (~50%) ( ). In a study of 365 AD patients, 22% of patients who reported pain rated pain intensity as 7 or greater ( ). In a different study of 144 AD patients, 14% of patients reported severe or very severe pain ( ).
AD patients often complain of both itch and pain, and itch and pain sensations were found to be highly correlated with one another ( ), suggesting a similarity between these phenomena. This chapter explains the neurosensory mechanisms of itch and pain in AD, focusing on neuronal sensitization, a phenomenon in which a minimal sensory stimulus results in an enhanced neural response ( ).
Pathogenesis of itch in atopic dermatitis and neuronal sensitization
Pathogenesis of itch in atopic dermatitis
Itch begins at the skin when unmyelinated, itch-selective C fibers are activated by pruritogen(s) in the epidermis and dermal-epidermal junction. Pruritogens are substances or molecules that have the potential to cause itch ( Table 16.1 ). These nerve fibers are categorized as histaminergic or nonhistaminergic depending on the receptors they express. Activation of these receptors results in the opening of ion channels, which leads to induction of an action potential. This signal is then transmitted to the spinal cord via the spinothalamic/spinoparabrachial tract, and is ultimately processed in the cerebral cortex of brain, which interprets and signals “itch” to the patient ( ). Note that histaminergic and nonhistaminergic nerve fibers transmit itch via separate neural tracts in the peripheral and central nervous system ( ).
| Category | Pruritogens |
|---|---|
| Neuropeptide |
|
| Protease | Tryptase, cathepsin S, kallikreins |
| Amines | Histamine, serotonin, bradykinin, endothelin |
| Cytokines |
|
| Lipid mediators | PGE2, phospholipase A2 |
Neuronal sensitization of itch
The perception threshold of itch is significantly lower in AD patients as compared with healthy controls. Itch neuronal sensitization is represented by two classically known phenomena: alloknesis and hyperknesis. Alloknesis is a phenomena in which normal or nonpruritic stimuli are perceived as itchy. Hyperknesis is excessive itch perception to pruritic stimuli ( ). Neuronal sensitization can occur due to various factors acting at the peripheral level (i.e., the skin) or at the central level (i.e., the brain or spinal cord). Peripheral sensitization is characterized by hypersensitivity of sensory neurons to itch stimuli caused by inflammatory mediators. Central sensitization is promoted by upregulation of itch-related receptors and molecules, dysfunction of the inhibitor circuits in the spinal cord, attenuation of descending inhibitory pathways in the spinal cord, and functional and structural changes in the brain ( ) ( Fig. 16.1 ).
The epidermis, barrier dysfunction, and neuronal sensitization
The role of barrier dysfunction in AD-associated itch is multifactorial. Cross-talk between nerve fibers may be an explanation ( ). Impaired barrier function can promote entry of pruritogens such as allergens and proteases into the skin. In addition, damage to the stratum corneum may lead to pruritogen release by keratinocytes ( ). The acidity of the stratum corneum is important for barrier formation and defense against bacteria, and skin pH in AD patients was shown to be elevated. High skin pH enhances the activity of proteases, which can act as pruritogens, thus contributing to itch in AD.
Nerve growth factor (NGF) is a neuropeptide that is secreted mainly by epidermal keratinocytes and promotes neural survival. Expression of NGF and its high-affinity receptor TrkA (tropomyosin receptor kinase A) is increased in AD. NGF may sensitize peripheral nerve endings through promotion of nerve sprouting and elongation ( ). NGF is also capable of increasing sensory nerve sensitivity to substance P and brain-derived neurotrophic factor (BDNF), which further contributes to the lowered itch threshold seen in AD patients.
Itch mediators in the skin and neuronal sensitization
Receptors
Unmyelinated C fibers transmit itch upon activation of specific receptors. The majority of these receptors are G protein–coupled receptors (GPCRs). A notable receptor within GPCRs is protease-activated receptor-2 (PAR2), which can be activated by various proteases such as tryptase (a secretary granule primarily by mast cells) and dust mites ( ). AD patients exhibited increased levels of tryptase in lesional skin and PAR2 in both lesional and nonlesional skin ( ). Administration of a PAR2 agonist caused prolonged itching in AD patients ( ).
Ion channels
When pruritogens encounter their receptors on C nerve fibers, calcium influx through specific ion channels is required to generate action potentials and propagate the itch signal. These ion channels include the transient receptor potential (TRP) family, which is a main component of all sensory perception, including itch, and voltage-gated sodium ion channels (Na v ).
TRP vanilloid 1 (TRPV1) exhibited increased expression in AD lesions, and its activation results in inflammation and pruritus. TRP ankyrin 1 (TRPA1) is involved in the transmission of histamine-independent itch. TRPA1 expression is enhanced in lesional skin of AD compared to healthy controls ( ). PAR2 can sensitize TRP channels, including TRPV1 and TRPA1 on peripheral nerve fibers, resulting in peripheral neuronal sensitization ( ).
TRPV1/TRPA1 activates Na v 1.7, a subtype of the Na v family. Na v 1.7 controls action potentials in itch mediation. Na v 1.7 mRNA expression is enhanced in pruritic skin lesion in AD ( ). In addition, a monoclonal antibody against Na v 1.7 attenuates itch sensation and pain ( ). Conversely, overactivity of Na v 1.7, which may be induced by skin inflammation, can lead to peripheral neuronal sensitization of itch.
Amines
Histamine is a prototype pruritogen that is involved in acute itch and released primarily by mast cells, basophils, and keratinocytes. However, psychophysiologic studies demonstrated that AD patients showed nonhistaminergic neuronal itch sensitization (but not histaminergic itch sensitization) ( ) and that antihistamines are not effective in treating chronic itch in AD. Thus nonhistaminergic causes are now considered to play a central role in AD itch ( ). Histamine may play a partial role in chronic itch of AD as studies involving blockers against H4R, a subtype of histamine receptor, have shown promising results ( ).
Furthermore, serotonin and bradykinin, molecules which normally function as endogenous algogens (pain-inducing agents), can cause neuronal sensitization and serve as potent nonhistaminergic pruritogens in lesional skin of AD.
Cytokines
Th2-related cytokines, such as interleukin-4 (IL4), IL13, IL31, and IL33, are known to be involved in the pathogenesis of AD and also reported to be involved in peripheral neuronal sensitization in AD. IL4 and IL13 are canonical Th2-related cytokines that help to drive AD ( ), and their expression is increased in lesional skin of AD. IL13 was shown to be a potent stimulator of itch in murine AD model and to strongly stimulate TRPA1 expression on nerve fibers ( ). In addition, IL4/IL13 can sensitize sensory neurons via IL4 receptor subunit α (IL4Rα). Dupilumab, an anti-IL4Rα monoclonal antibody, can greatly improve pruritus in AD even before the improvement of skin symptoms, indicating a promoting role of IL4/IL13 in the development of itch ( ).
IL31 is generated mainly by activated Th2 cells and has been coined the “itchy cytokine.” IL31 receptor transcripts are abundantly expressed in dorsal root ganglia ( ). Lesional IL31 expression was increased in AD compared with controls. Serum IL31 levels were correlated with disease activity in AD. IL31 provokes pruritus through directly stimulating sensory neurons and may do so indirectly through its interaction with keratinocytes ( ). Additionally, IL31 was also shown to induce sensory nerve elongation and branching, resulting in peripheral neuronal sensitization ( ). Nemolizumab, an anti-IL31 receptor A monoclonal antibody, dramatically improves itch in AD patients.
IL33 is a keratinocyte-derived cytokine. Lesional IL33 levels were increased in AD patients. Serum IL33 levels were correlated with disease severity ( ). IL33 can trigger Th2-associated inflammation and directly stimulate itch sensory neurons ( ). IL33 also can contribute to peripheral neuron sensitization in immune inflammation responses ( ).
Thymic stromal lymphopoietin (TSLP) and IL17A are also implicated in AD-associated itch. They are reported to play a role in pain neuronal sensitization and may be involved itch neuronal sensitization; however, there is no direct evidence for this. TSLP is a Th2-related cytokine secreted by keratinocytes partially via PAR2 and is able to directly stimulate itch sensory neurons via TRPA1 ( ). IL17A is a Th17-related cytokine involved in the acute phase of AD. mRNA expression of IL17A and its related cytokine IL23A is enhanced in itchy AD lesions compared with nonitchy skin ( ).
Neuropeptides
Aside from NGF, substance P is another tachykinin neuropeptide implicated in AD itch. Plasma substance P levels were higher in AD patients than in healthy controls and significantly correlated with disease activity ( ). Substance P initiates itch once bound to its receptor neurokinin receptor-1 (NK-1) on various cells (e.g., mast cells), resulting in the release of histamine and nonhistaminergic itchy mediators such as leukotrienes (LTs) and prostaglandins (PGs). These mediators are involved in neuronal sensitization (see Chapter 2 ).
Lipid mediators
Production of PGs, including PGE2, is increased in AD lesions. PGs can directly stimulate their receptors expressed on sensory nerve fibers. In addition, activated C fibers in response to PGs are capable of producing neurogenic inflammation via release of neuropeptides, including substance P. In this way, PGs can be involved in peripheral neuronal sensitization to both itch and pain ( ).
Itch processing in the spinal cord and neuronal sensitization
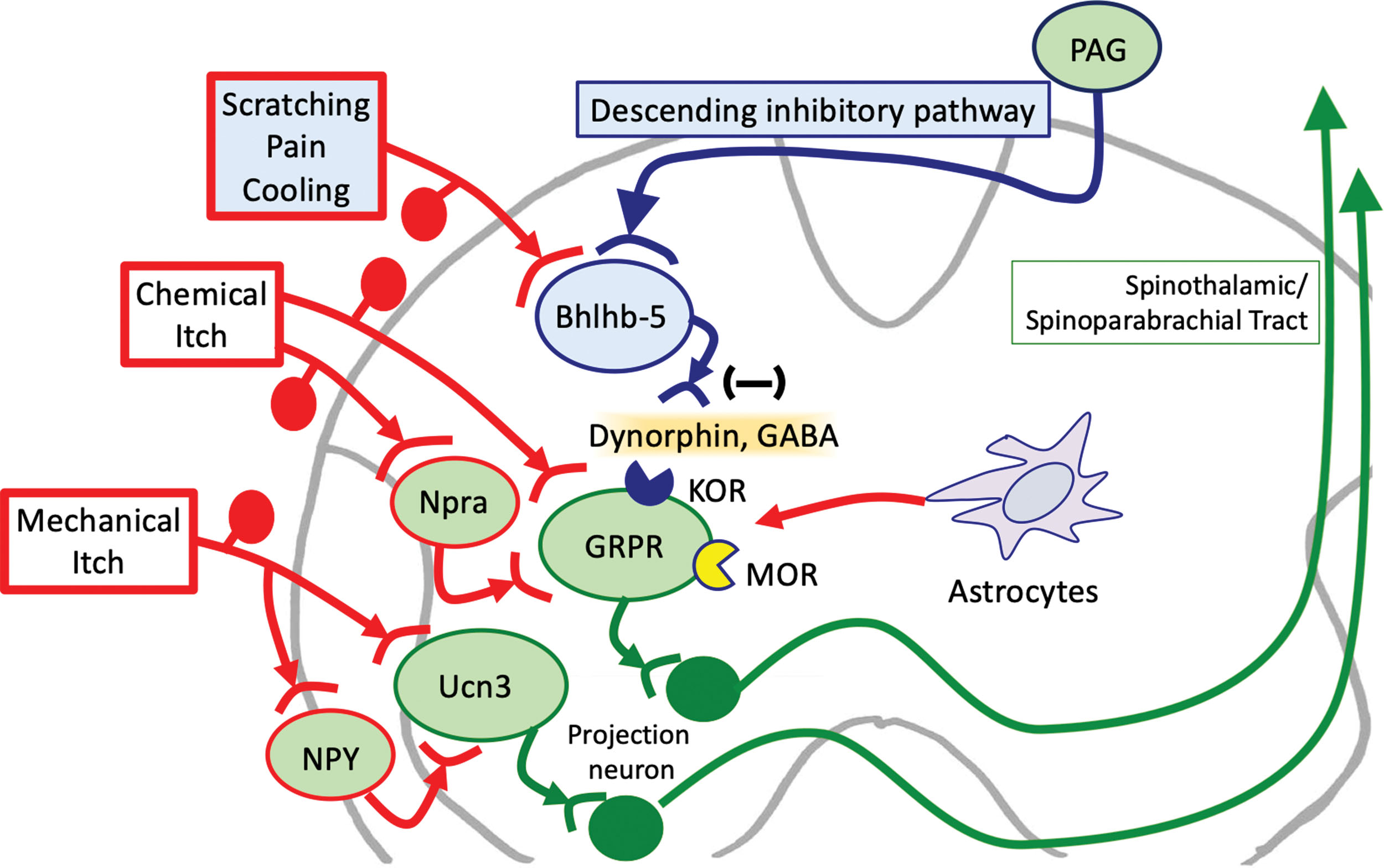
Itch signals are modulated by interneurons in the spinal cord ( Fig. 16.2 ). Chemical itch is transmitted through gastrin-releasing peptide receptor (GRPR) positive neurons. The transmission of chemical itch through GRPR+ neurons can be inhibited by helix-loop-helix family member B5 (Bhlhb5)–positive inhibitory interneurons upon their secretion of several molecules, including γ-amino butyric acid (GABA). Mechanical itch is conveyed by another itch-related circuit in the spinal cord, which involves urocortin 3 (Ucn3)–positive excitatory interneurons ( ). Ucn3+ interneurons are inhibited by neuropeptide Y (NPY)–positive neurons. Dysfunction of inhibitory neuronal circuits (Bhlhb5+ neurons and NPY+ neurons) can lead to enhanced neural sensitization.