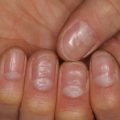
Fig. 20.1
Urticaria (Courtesy of Adelaide A. Hebert, MD)
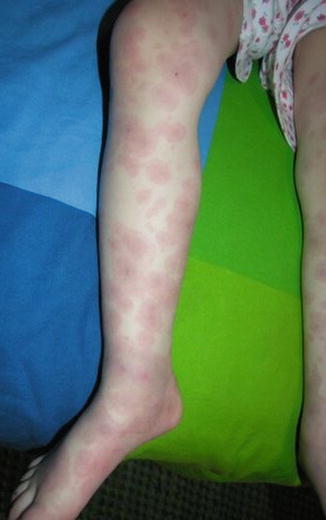
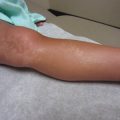
Fig. 20.2
Urticaria (Courtesy of Adelaide Hebert,MD)
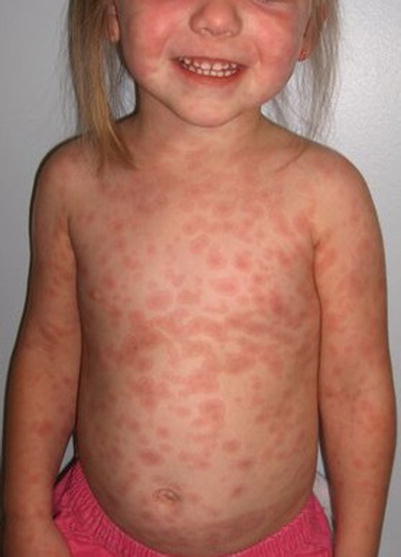
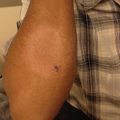
Fig. 20.3
Urticarial multiforme (Courtesy of Adelaide Hebert, MD)
Management Strategies
Differential diagnoses include viral exanthema, contact dermatitis, drug eruptions, insect bites, and erythema multiforme. Acute urticaria in children has been largely seen following a viral or bacterial infection. Many children receive antibiotics after a bacterial infection and then develop urticaria, rendering it difficult to attribute cutaneous findings to an infection versus a drug reaction. In angioedema without urticaria, the clinician should think of other disorders such as drug-induced angioedema, idiopathic angioedema, or hereditary and acquired C1 inhibitor deficiency. Initial management should be targeted on relieving pruritus and angioedema, if present. Two-thirds of cases of urticaria are self-limiting [1]. For this reason, the treatment options have not been extensively studied for acute cases. Many of the treatments are anecdotal recommendations from studies of chronic urticaria.
Specific Investigations Recommended
For diagnosis |
Clinical |
CBC with differential, urinalysis, ESR, LFTs |
Allergy-specific IgE antibody |
A diagnosis of urticaria can be made clinically with a good history and cutaneous findings of transient, pruritic wheals. To address the underlying cause, a basic laboratory work-up may be conducted to include a cell blood count (CBC) with differential, urinalysis, erythrocyte sedimentation rate (ESR), and liver function tests (LFTs). If a certain allergy is suspected, an allergy-specific immunoglobulin E (IgE) antibody may be performed.
Biopsy is not indicated unless the case is atypical. Histopathology demonstrates a mild, lymphocytic perivascular infiltrate with marked dermal edema [2].
Table 20.1
First line therapies
Second-generation H1 antihistamines | B |
Combining H1 and H2 antihistamines | B |
Most commonly, the recommended therapy will be second-generation H1 antihistamines, as they are non-sedating [3]. Doses can be prescribed as following: cetirizine 5 to 10 mg daily for children >6 years; 2.5 to 5 mg for children 2–5 years; 2.5 mg for children 6 months to 2 years. Alternatives include levocetirizine, loratadine, desloratadine, and fexofenadine. Combining H1 and H2 antihistamines for patients with acute urticaria has been shown to produce a better response [4]. than use of H1 antihistamines alone (in some published studies). H2 antihistamines include ranitidine, nizatidine, and famotidine.
In an emergency room setting, parenteral dosing of first-generation H1 antihistamine is available: diphenhydramine 0.5–1.25 mg/kg (up to 50 mg per dose) IV/IM every 6 h as needed or hydroxyzine 0.5–1 mg/kg (up to 50 mg per dose) IM every 6 h as needed.
Table 20.2
Second line therapies
First-generation H1 antihistamines | D |
Systemic Glucocorticoids | E |
First-generation H1 antihistamines are still used to treat urticaria. despite the fact that this class of medications can they cross the blood-brain barrier and cause a sedating side effect.
Table 20.3
Third line therapies
Omalizumab | A |
Ecallantide (Kalbitor) ®a | A |
Antileukotriene agents | B |
Cyclosporine | D |
Omalizumab, a recombinant humanized monoclonal anti-IgE antibody, has been used to treat patients with recurrent angioedema and chronic urticaria within 1–2 weeks [5]. A randomized, double-blind clinical trial was conducted on patients ages 12–75 who continued to be symptomatic while on antihistamines. Subcutaneous omalizumab was effective in decreasing pruritus [6]. Ecallantide (Kalbitor®), a kallikrein inhibitor, blocks the production of bradykinin and therefore its effects. Multiple randomized, double-blind clinical trials were performed in different age groups which showed that ecallantide was effective in relieving acute hereditary angioedema attacks. This medication is administered subcutaneously in a hospital setting or infusion center, to monitor for possible side effects. In clinical trials, 4 % of patients experienced anaphylaxis [7]. Antileukotriene agents (montelukast) have not been studied in acute cases; however, this agent has been used in chronic cases of urticaria. These medications are not demonstrably more effective than combining H1 and H2 antihistamines. Cyclosporine has been used with to treat chronic disease with positive results; however, patients are still susceptible to relapses [8].
Drug Hypersensitivity Reaction
Clinical Features
Drug hypersensitivity reactions, also known as exanthematous drug eruptions, are type IV, T-cell mediated immune reactions that are the most common type of adverse drug reaction. Patients develop erythematous macules or small papules that appear 7–14 days after drug exposure [9]. This classic drug-induced skin eruption predominantly involves the trunk and proximal extremities. Typically, there is no mucosal membrane involvement. Cutaneous drug reactions are morbilliform 95 % of the time, with 5 % being urticarial (Figs. 20.4 and 20.5). Synonyms include morbilliform or maculopapular drug eruption.
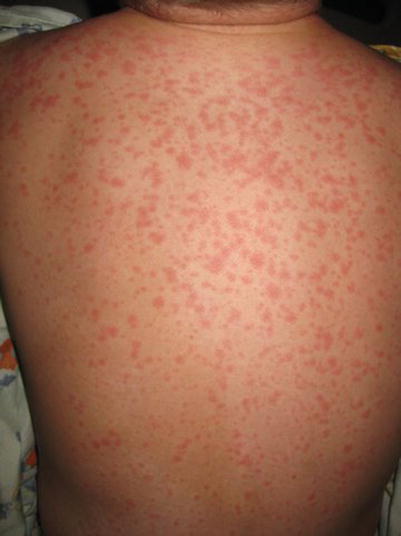
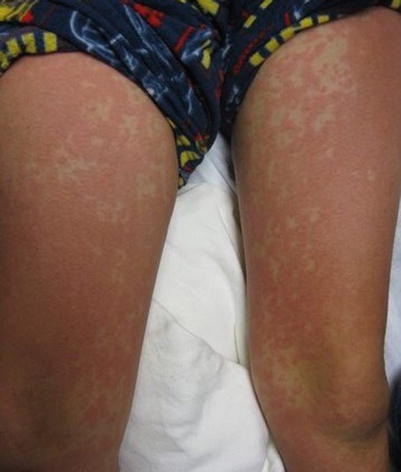
Fig. 20.4
Drug hypersensitivity reaction (Courtesy of Adelaide Hebert, MD)
Fig. 20.5
Drug hypersensitivity reaction (Courtesy of Adelaide Hebert, MD)
Management Strategies
Differential diagnosis includes viral and bacterial exanthems, contact dermatitis, psoriasis, and other eruptions associated with systemic diseases. The cutaneous signs of a drug eruption are self-limiting. Treatment is targeted toward symptom relief such as pruritus. In severe disease with mucosal or genital involvement, suspect drug rash with eosinophilia and systemic symptoms (DRESS), Stevens-Johnson syndrome (SJS), or toxic epidermal necrolysis (TEN). A thorough medical history is imperative in order to identify a chronological relationship between drug exposure and symptoms. Next, discontinuation of the offending agent (when deemed feasible) is advised. Even with discontinuation, cutaneous findings and symptoms may persist and take up to 7–14 days, or longer, to begin to improve. In patients with serious drug eruptions who take several medications, the nonessential drugs should be discontinued, if possible. Laboratory tests are only necessary if the diagnosis is not clear from the history and the clinical correlation.
Specific Investigations Recommended
For diagnosis |
CBC with differential |
LFT and Renal panel |
ANA (antinuclear autoantibody) |
Skin biopsy |
For treatment |
Patch Testing 1–6 months after symptoms |
CBC with differential is performed to identify eosinophilia, changes in white blood cell count, and alteration of platelets. LFT and renal panel can be performed to identify systemic involvement, especially if DRESS is suspected. If an autoimmune connective tissue disease is in the differential, ANA and other studies directed at discovery of the suspected autoimmune disorder should be performed.
A skin biopsy is often not necessary, and only warranted if a severe hypersensitivity reaction is noted. Biopsies may be considered in patients with systemic symptoms such as fever, signs of systemic involvement, or mucous membrane involvement, or if the cutaneous symptoms evolve to erythroderma, blistering, or pustule formation. The histopathology of most drug eruptions display interface dermatitis with dyskeratotic keratinocytes along the dermoepidermal junction and scattered eosinophils.
Table 20.4
First line therapies
Avoid offending agent | D |
Topical corticosteroids | B |
Oral antihistamines | B |
Patients complain of moderate to severe pruritus; therefor, topical corticosteroids with medium to high potency are suggested (group 1–3 on the corticosteroid potency chart). In order of increasing strength, these include: triamcinolone acetonide 0.5 % cream (Triderm), fluocinonide 0.05 % cream (Lidex-E), betamethasone dipropionate 0.05 % cream (Diprolene), or clobetasol propionate 0.05 % cream (Temovate). A wide variety of topical steroids are available. A list of topical steroids in a wide variety of formulations (creams, ointments, foams, gels, solutions, tape) separated by strength and class is available on the National Psoriasis Foundation website at: https://www.psoriasis.org/about-psoriasis/treatments/topicals/steroids.
Antihistamines (sedating and non-sedating) can be used to treat pruritus systemically. These include diphenhydramine, hydroxyzine, and non-sedating cetirizine (Table 20.5). Hydroxyzine and cetirizine are administered until the pruritus subsides.
Table 20.5
Antihistamine dosing
Medication | Age | Dose |
|---|---|---|
Diphenhydramine (Benadryl) | 2–5 years | 6.25 mg every 4–6 h |
6–11 years | 12.5–25 mg every 4–6 h | |
≥12 years | 25–50 mg every 4–6 h | |
Hydroxyzine (Atarax, Vistaril) | <6 years | 1–2 mg/kg/day divided into 6–8 h |
≥6 years | 12.5 to 25 mg every 6–8 h | |
Cetirizine (Zyrtex, Reactine) | 6–12 months | 2.5 mg daily |
12 months to <2 years | Start at 2.5 mg daily, may increase to 2.5 mg twice daily or 5 mg daily | |
2–5 years | 2.5 mg daily, may increase to 2.5 mg twice daily or 5 mg daily | |
≥6 years | 10 mg daily |
Table 20.6
Second line therapies
Systemic corticosteroids | E |
Systemic corticosteroids are not recommended unless the patient has systemic involvement or severe cutaneous symptoms [10]. In these cases, a short course of prednisone 1–2 mg/kg/day may be beneficial.
Table 20.7
Third line therapies
Desensitization | B |
Desensitization can be used to decrease the allergic response to a necessary medication. In patients who do not require a certain medication or are able to substitute for another medication, then desensitization is not necessary. However, in the subset of the population who have an exanthematous drug eruption to a medication that is necessary, this is a viable option [11, 12]. Desensitization is typically carried out in a hospital setting, with adequate support to handle any urgent medical need should one arise during the process of desensitization.
Fixed Drug Eruption
Clinical Features
A fixed drug eruption (FDE) is a specific cutaneous drug reaction that recurs in the same area when re-exposed to an offending medication. Acute FDE typically presents as a single plaque or a small cluster of dusky red, violaceous plaques that appear 30 min to 8 h after drug administration (Fig. 20.6). Sometimes this cutaneous finding can take several days to evolve. Typical plaques of a fixed drug eruption are well-demarcated, round to oval, and with edematous skin changes with or without vesiculation (Fig. 20.7). These fixed skin lesions can occur anywhere on the body, but are seen most commonly on the lips, genitalia, perianal area, hands, feet, and/or any site of previous trauma [13, 14]. Mucosal involvement can be a manifestation of a fixed drug eruption. Upon resolution of the fixed drug eruption, plaques can leave behind post-inflammatory hyperpigmentation. Rarely, patients can have atypical variants of severe FDE, which include multiple plaques, non-pigmenting lesions, or generalized bullous lesions that may appear similar to more serious diseases such as Stevens-Johnson syndrome (Fig. 20.8).
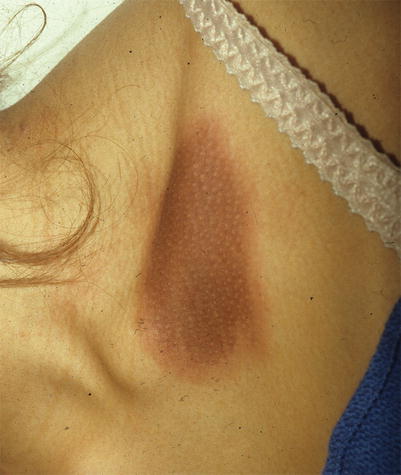
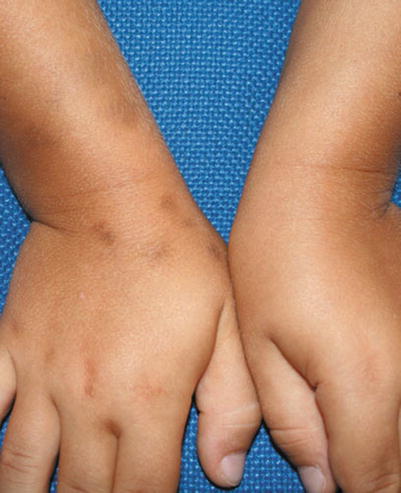
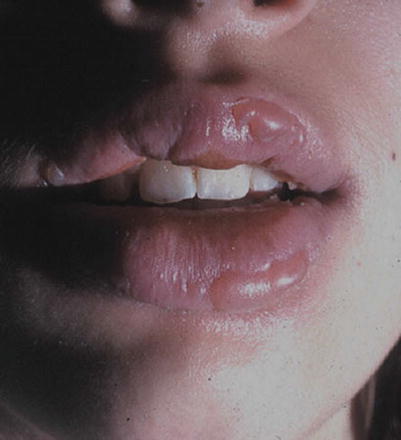
Fig. 20.6
Fixed drug eruption (Courtesy of Adelaide Hebert, MD)
Fig. 20.7
Fixed drug eruption (From Bonifazo E. Differential Diagnosis in Pediatric Dermatology. In: Allergic Diseases. Springer; 2013. p. 95. Used with kind permission from Springer Science and Business Media)
Fig. 20.8
Fixed drug eruption (From Bonifazo E. Differential Diagnosis in Pediatric Dermatology. In: Allergic Diseases. Springer; 2013. p. 95. Used with kind permission from Springer Science and Business Media)
The hallmark of FDE is that lesions reappear at the same site when patients are re-exposed to the offending agent. New lesions can occur in new sites as well. Cross-reactivity is possible from medications that have a similar chemical structure than the offending medication.
Pruritus is a common finding in fixed drug eruptions, although other systemic symptoms are typically absent. In the pediatric population, FDEs account for 14–22 % of cutaneous drug reactions [15]. The localized response of FDE is attributed to intraepidermal CD8+ T cells [16]. When these cells are activated, they release interferon-γ and cytotoxic granules [17]. The most common culprits of FDEs include antibiotics (trimethoprim, sulfamethoxazole, tetracycline, and penicillins), NSAIDs, acetaminophen, phenolphthalein, barbiturates, and antimalarials.
Management Strategies
Differential diagnosis includes erythema multiforme (EM) and SJS/TEN for the generalized bullous FDE variant. Diagnosis is made after history and identification of lesion morphology. Having a recurrence of the fixed dusky plaque at the same anatomic site is very supportive for the diagnosis of FDE. Once the offending agent is identified, immediate discontinuation of the offending agent is recommended. After this is done, the classic skin lesion will persist for 7–10 days, leaving behind a patch of post-inflammatory hyperpigmentation.
Specific Investigations Recommended
For diagnosis |
Clinical |
Biopsy |
Oral challenge |
Patch testing |
Diagnosis is typically clinical; however further investigation is warranted when this is not obvious. FDE on histology will display hydropic degeneration of the basal layer, pigmentary incontinence, dyskeratotic cells, and dermal lymphocytic infiltrates.
An oral challenge test or patch testing can be performed to identify the offending agent. Oral challenge test is not recommended if the patient has ever developed a generalized FDE. In a study of 450 patients who received the oral challenge test with the medication believed to be causing their symptoms, 10 % developed pruritus, 0.9 % had a fever, and 0.7 % developed generalized urticaria. No severe reactions were noted [18]. During an oral challenge test, patients do not get tested with the full dose of the presumed offending medication. In patients with a history of generalized FDE, patch testing is a valid option. There are different methods to perform a patch test. In one study, 1–5 % of the therapeutic dose combined with a diluting agent such as white soft paraffin was applied to the patients’ skin. The suspected medication is applied to an old FDE lesion to elicit a local reaction as well as to normal tissue to compare. If no reaction is witnessed, the dose can be titrated upwards. As this testing methodology is not considered to be systemic, this test is considered safe for patients with previous history of FDE. More methods of performing a patch test are to mix the implicated drug with petrolatum or dilute in water at 10–20 % concentration and then apply it to the skin. A reaction is considered positive when erythema occurs within 24 h and lasts at least 6 h [19].
Table 20.8
First line therapies
Discontinue offending drug | D |
Topical corticosteroids | B |
Systemic antihistamines | B |
The first step in treatment of FDE is to withdraw and avoid the offending agent, and avoid all drugs that are chemically related. Patients should be provided with a list of medications to avoid that includes the generic name, trade names, and medications that cross-react. Similar to drug hypersensitivity reactions, FDE is self-limiting once the offending agent is discontinued. Therapy is targeted to treat pruritus and educate the patient regarding both what to expect during the resolution of the FDE and how to potentially avoid this skin reaction in the future. For small or single FDE lesions, topical corticosteroids (group 1–3) can be applied daily for 7–10 days. For diffuse pruritus, oral H1 antihistamines are prescribed (Table 20.5).
Table 20.9
Second line therapies
Systemic corticosteroids | – |
In cases of generalized FDE, or when systemic systems are present, a short course of moderate dose systemic corticosteroids may be beneficial. This has yet to be proven in randomized, double-blind trials; however, numerous clinicians attest to the effectiveness of this therapeutic approach through clinical experience. The administration of prednisone (Rayos, Sterapred) 0.5–1 mg/kg/day for 3–5 days can alleviate pruritus and potentially hasten the resolution of the FDE.
Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis
Clinical Features
SJS and TEN (erythema multiforme major) are severe mucocutaneous adverse reactions that are part of the same disease continuum, and are distinguished by the disease severity and extent. Patients develop fever, cutaneous discomfort, and extensive epidermal detachment and necrosis. This skin reaction is typically attributed to a medication or an infectious agent. Bullous erythema multiforme (EM) is a milder reaction with only cutaneous symptoms. This is characterized by flat or slightly raised target lesions with epidermal involvement less than 10 % body surface area (BSA).
In SJS, patients develop widespread macules or target lesions also less than 10 % BSA, and the mucous membranes are involved in over 90 % of patients with presentation in at least two distinct sites (oral, ocular, and genital). When epidermal necrosis and detachment is between 10 % and 30 % BSA, this is diagnosed as an overlap SJS-TEN, and when BSA is greater than 30 %, TEN is diagnosed (Fig. 20.9). TEN has the same incidence of mucosal involvement as SJS, with over 90 % of patients having the mucous membranes affected. As SJS and TEN are in the same continuum, these two potentially life-threatening cutaneous disorders will be discussed together.
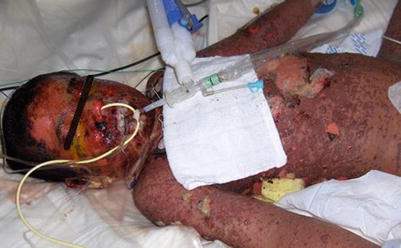
Fig. 20.9
Toxic epidermal necrolysis (Courtesy of Adelaide Hebert, MD)
Patients typically complain of fever and flu-like symptoms preceding the mucocutaneous symptoms by 1–3 days. Physicians should suspect SJS/TEN in patients who have fever, skin tenderness, blistering, and mucosal inflammation [20]. Cutaneous findings begin with ill-defined, coalescing erythematous macules with pruritic centers. Lesions start on the face and thorax before spreading to rest of body. Scalp, palms, and soles are rarely involved. As disease progresses, vesicles and bullae form and skin begins to slough. Patients will have a positive Nikolsky sign. The skin surface will resemble that of a burn patient. Most patients develop painful, hemorrhagic erosions of the oral mucosa and vermillion border (Figs. 20.10, 20.11, and 20.12)
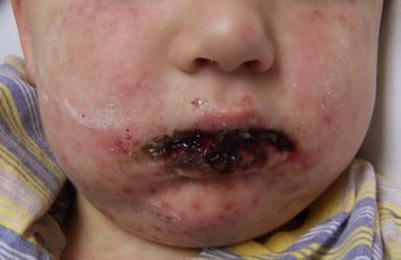
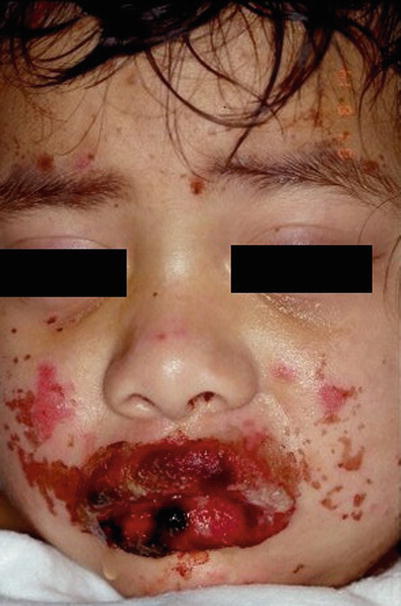
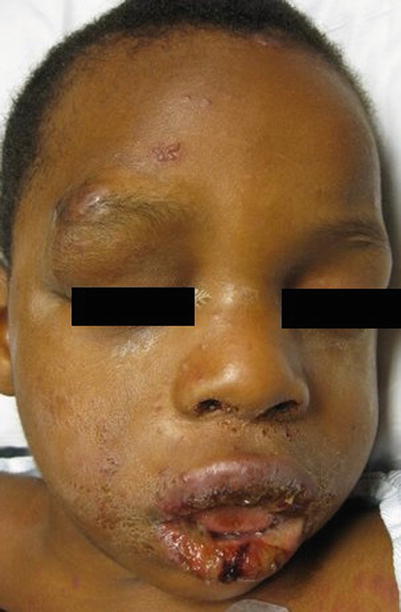
Fig. 20.10
Steven Johnson Syndrome (Courtesy of Adelaide Hebert, MD)
Fig. 20.11
Steven Johnson Syndrome (Courtesy of Adelaide Hebert, MD)
Fig. 20.12
Steven Johnson Syndrome secondary to Herpes Simplex Virus infection (Courtesy of Adelaide Hebert, MD)
Malnutrition and dehydration can develop, due to painful stomatitis and mucositis. Many affected patients also develop severe conjunctivitis with purulent discharge. Amongst survivors, long-term ocular complications such as corneal damage or conjunctival scarring puts them at risk for vision loss. Pharyngeal mucosa and urogenital involvement is common [21].
Medications attributing most in pediatric patients include sulfonamide antibiotics, phenobarbital, carbamazepine, lamotrigine, and acetaminophen (Paracetamol) [22]. While medications are the most likely cause of these conditions, greater than 25 % of pediatric cases cannot be clearly linked to a medication. The most common infectious causes are Mycoplasma pneumonia (especially if fever, cough, and mucositis is present) and cytomegalovirus [23]. On very rare occasions, vaccines have been known to cause SJS and TEN [24].
Medications stimulate the immune system by binding to major histocompatibility complex I (MHC-I) and T-cell receptors. This immune response leads to clonal expansion of cytotoxic T-cells that are drug-specific. These cells kill keratinocytes directly, and ultimately recruit cells that release cytokines.
Management Strategies
All suspected cases of SJS or TEN should be considered for hospital/intensive care unit admission. Once the diagnosis is verified, severity should be assessed and management initiated, including placement of the patient on a non-stick bedding and dressing such as ExuDry®. Differential diagnosis for SJS/TEN includes: EM, erythroderma (due to radiation toxicity or other causes), erythematous drug eruptions, acute generalized exanthematous pustulosis, phototoxic eruptions, and Staphylococcal Scalded Skin Syndrome. The management of SJS/TEN starts with immediate removal of the offending agent, IV antibiotics (treatment or prophylaxis), hospitalization, nutritional and supportive care, and wound care.
Adults have a higher mortality rate than children when afflicted with TEN; however 50 % of children have long-term sequelae as a result of this severe cutaneous disease. In a series of 55 children with SJS/TEN, 10 had recurrence up to 7 years after their initial episode. Recurrence was mostly attributed to Mycoplasma pneumonia infection, herpes simplex virus, or antiepileptic drugs [25]. This demonstrates that patients can have a long-term immunosensitivity to certain medications, and should indefinitely be classified as hypersensitive/allergic to offending agents and medications with the same chemical structure.
There is a wide array of sequelae that can develop after recovery from SJS/TEN. Patients can develop irregular pigmentation patterns, alopecia, abnormal nail growth, xerostomia, gingival synechiae, long-term vulvovaginal dryness, urinary retention, chronic bronchitis/bronchiolitis, and multiple ophthalmologic problems such as photophobia or visual impairment.
Specific Investigations Recommended
For diagnosis |
SCORTEN |
CBC, LFT, Renal panel |
Cultures |
Mycoplasma pneumoniae serology |
Biopsy |
For treatment |
Cultures (wound and lines) during hospitalization |
Due to the severity of this group of skin disorders, basic laboratory work-up and cultures are necessary, both initially and as the patient is managed over the course of their disease progression. Patients develop anemia and lymphopenia. One-third of patients have neutropenia. which is correlated with a poor prognosis [26]. Major fluid loss can lead to increased BUN, hyperglycemia, hypoalbuminemia, and electrolyte imbalance. Mild elevations in serum aminotransferases of about two to three times the normal limit are present in half of TEN patients. There is a high risk of bacterial superinfection and sepsis; therefore, cultures should be taken from the blood, cutaneous wounds, and mucosal wounds. Mycoplasma pneumonia serology is also obtained in the early stage of the disease and 3 weeks following.
A skin biopsy should be performed in most cases to verify the suspected diagnosis. The hallmark of SJS/TEN on histopathology is partial or full-thickness keratinocyte necrosis of the epidermis, typically without inflammation [27].
Table 20.10
First line therapies
Immediate discontinuation of offending agent | B |
Wound care | B |
Supportive care | – |
Transfer to burn center | B |
A 10-year observational study of 203 adult patients with SJS or TEN concluded that early withdrawal of the offending agent decreased mortality, especially if withdrawn before the presence of blistering [28]. The administration of supportive care, inclusive of wound care, fluid and electrolyte management, nutritional support, and pain control, are essential to assure an optimized outcome. Even with meticulous care, a large number of severely affected patients succumb to adverse events from the disease. Multiple studies have been conducted comparing different wound care techniques: debriding wounds versus leaving detached skin intact, or using nonadherent dressings with silver versus petrolatum. None of the studies found one technique to be superior to another in regard to rates of survival or reepithelialization.
Although controversial to some, prognosis is improved when patients with severe cutaneous and systemic symptoms are transferred early to burn centers [29]. Room temperatures should be raised to prevent excessive caloric loss due to uncontrolled muscle shaking to help maintain core body temperature. Nasogastric feeding may be necessary to endure adequate calories for the stress of the disease and to promote cutaneous recovery. Bronchial injury and hypersecretions should be managed by a trained respiratory therapist, with chest physical therapy and pulmonary toileting. Extra care should be taken when passing the feeding tube, to avoid further damage to affected mucous membranes.
All patients who are admitted to the hospital should get immediate eye exams by an ophthalmologist because the ocular inflammation and damage can develop quickly. Genitourinaryexaminations should be conducted for both males and females to evaluate for complications such as phimosis, urethral strictures, or vaginal or labial adhesions. In patients with intravaginal ulcers, a moderate-potency topical corticosteroid gel can be applied intravaginally twice daily. Topical or systemic antifungals can be used concomitantly to prevent vaginal candidiasis.
Table 20.11
Second line therapies
Systemic antibiotics | E |
Once cutaneous symptoms are present, care should be provided in a manner similar to the technique used in a burn unit. Skin and line cultures should be considered every 48 h. Systemic antibiotics are not necessary, unless the patient displays symptoms such as fever or general deterioration with evidence of infection. Infections with gram negative rods, especially Pseudomonas, can be especially problematic and should be dealt with promptly.
Table 20.12
Third line therapies
Intravenous immunoglobulin | D |
Systemic corticosteroids | D |
Hemoperfusion | C |
Cyclosporine | E |
There have been many retrospective and a few controlled studies evaluating the use of intravenous immunoglobulin (IVIG) and systemic corticosteroids in adults. Overall, the data shows decreased rates of mortality with both medications when used alone or together. In an adult meta-analysis, IVIG doses ≥2 g/kg significantly decreased mortality in both SJS and TEN [30]. In pediatric patients, the use of IVIG and/or systemic corticosteroids has been reported in case studies and still used anecdotally based on the data in adult studies. Given the side effects of these medications, there use can be controversial. Theoretically, they increase the risk of sepsis and decrease the rate of epithelialization. If considering these agents, therapeutic interventions should be given made on a case-by-case basis.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


