The histiocytoses are a broad group of disorders characterized by an abnormal proliferation of the histiocyte, a type of progenitor cell in the bone marrow ( Box 10-1 ). Some clinically relevant types of histiocytes include the Langerhans cell, the dermal dendrocyte, and cells of mononuclear cell/macrophage lineage. Malignant disorders to be discussed include hematologic malignancies (including leukemia, lymphoma, and Hodgkin disease), neuroblastoma, and some sarcomas.
Langerhans cell histiocytosis
Juvenile xanthogranuloma
Xanthoma disseminatum
Benign cephalic histiocytosis
Necrobiotic xanthogranuloma
Generalized eruptive histiocytoma
Progressive nodular histiocytoma
Indeterminate cell histiocytosis
Multicentric reticulohistiocytosis
Sinus histiocytosis with massive lymphadenopathy (Rosai–Dorfman disease)
Hemophagocytic syndromes
Malignant histiocytic syndromes
Langerhans Cell Histiocytosis
Langerhans cell histiocytosis (LCH) is the term now used to describe a disorder characterized by infiltration of Langerhans cells into various organs of the body. Older synonymous terms, which are now largely obsolete or unnecessary, include histiocytosis X, eosinophilic granuloma, Letterer–Siwe disease, Hand–Schüller–Christian disease, and Hashimoto–Pritzker disease. The term histiocytosis X was coined by Lichtenstein in 1953 to identify three related clinical entities of unknown etiology and characterized by histiocyte proliferation. This classification included the triad of Letterer–Siwe disease, Hand–Schüller–Christian disease, and eosinophilic granuloma. The X in this original nomenclature was used to denote the unknown derivation of the histiocyte involved in this disorder. Ultrastructural studies eventually confirmed the relationship of these three different presentations by showing the Langerhans cell to be the proliferative cell in each of them. LCH may occur at any age, from newborn to elderly, although the peak incidence appears to be between the ages of 1 and 4 years.
Langerhans cells are derived from the bone marrow and are a type of dendritic cell found primarily in the epidermis (as well as mucosal epithelia, thymus, esophagus, and lung). They are involved in antigen presentation for the skin- and mucosa-associated immune systems and are identified by strong staining with S100 (a neuronal protein) and cluster designation (CD) la (a cell surface marker). Langerhans cells also have a characteristic organelle, the Birbeck granule, which is visible on electron microscopy. The function of this organelle remains unknown. Immunohistochemical demonstration of langerin (CD207), a mannose-specific lectin found in association with Birbeck granules, has also been demonstrated useful for diagnostic confirmation of LCH.
Although numerous etiologies have been proposed for LCH, the pathogenesis remains obscure. Hypothetic causes include somatic mutations, infection (especially viral), immune or cytokine dysregulation, and programmed cell death (apoptosis). Whether LCH is a neoplastic versus a reactive disorder also remains debated. The cells in LCH have been demonstrated to be clonal, and such monoclonality has been demonstrated both in multisystem disease and with solitary organ involvement. Although this clonality suggests that LCH may be a neoplastic process with variable biologic behavior, the exact significance and implications remain controversial. There are arguments both in favor of and against LCH being a neoplastic process.
LCH may involve multiple organ systems, the most common being the skin and the bones. Table 10-1 lists the spectrum of organ involvement. The older classification system was based on the involved organ systems such that eosinophilic granuloma referred to localized bone disease; Hand–Schüller–Christian disease the multifocal triad of bone (usually skull) lesions, exophthalmos, and diabetes insipidus (DI); and Letterer–Siwe disease, the acute or subacute disseminated form of the disease. Under the more modern classification, LCH is the umbrella term for the disease with notation made of the various organ systems involved. Patients may have unifocal, multifocal, or disseminated disease. In general, patients with widespread, multiorgan involvement have the poorest prognosis, and those with isolated bone LCH have the best prognosis.
| Organ | Comment |
|---|---|
| Skin | Often the initial presenting sign |
| Bone | Painful swelling common; incidence: skull > long bones > flat bones (ribs, pelvis, vertebrae) |
| Lymph nodes | Cervical most common |
| Liver | |
| Spleen | |
| Lungs | Diffuse micronodular pattern on radiography |
| Gastrointestinal tract | |
| Thymus | |
| Bone marrow | Pancytopenia portends a poor prognosis |
| Gingivae, buccal mucosa | Swelling, erythema, erosions, petechiae |
| Kidney | |
| Endocrine glands | Diabetes insipidus most common |
| Central nervous system |
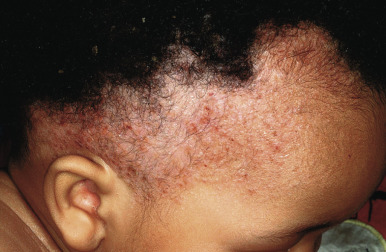
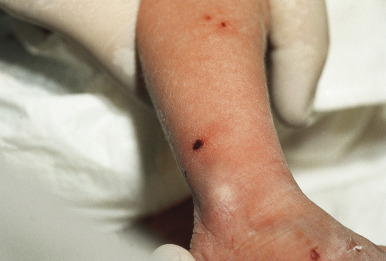
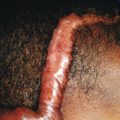
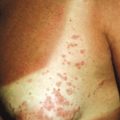
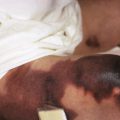
Cutaneous involvement is very common in LCH and is often the presenting complaint. The spectrum of skin findings is listed in Table 10-2 . The most classic presentation is that of a seborrheic dermatitis-like eruption with prominent involvement of the scalp, posterior auricular regions ( Fig. 10-1 ), perineum, and axillae. The rash tends to be resistant to standard therapy, which is an important clue that should prompt consideration of the diagnosis. Erythematous, red-brown papules are often seen, especially on the scalp and in flexural areas, and may have secondary erosion, hemorrhage, or crusting ( Fig. 10-2 ). Crusted papules on the palms and/or soles ( Fig. 10-3 ) are another important feature, especially in infants in whom the diagnosis of scabies has been excluded. Although this finding has traditionally been felt to portend a poor prognosis, this observation has not been validated.
| Presentation | Comment |
|---|---|
| Scaly red-brown papules | Especially in scalp, flexural areas; may have associated crusting; may be umbilicated or lichenoid; palm and sole involvement common |
| Erythematous, scaly dermatitis | May simulate seborrheic dermatitis |
| Erosion or ulceration | Common secondary finding, especially in fold areas |
| Petechiae, hemorrhage | With or without associated thrombocytopenia |
| Vesiculopustular lesions | Most common in neonates; may simulate congenital varicella or herpes; often have hemorrhagic crusts |
| Red nodules or plaques | Less common |
| Granulomatous plaques | Rare |
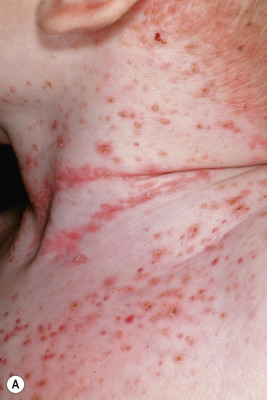
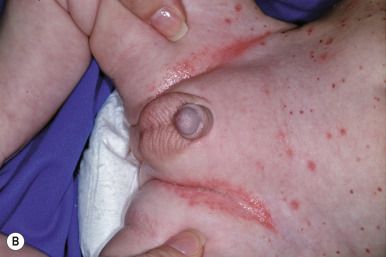
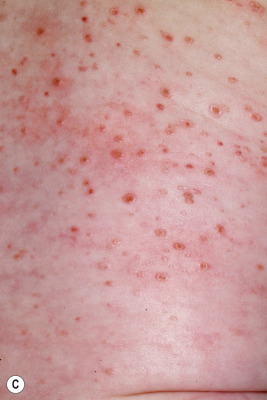
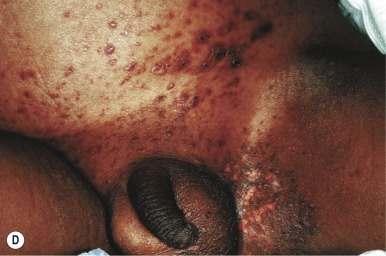

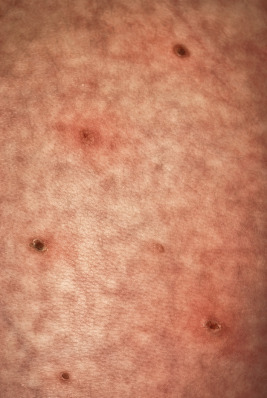
In neonates with LCH, vesiculopustular lesions ( Figs. 10-4 and 10-5 ) tend to predominate and may be misdiagnosed as congenital varicella or herpes. These lesions may become hemorrhagic or crusted. Petechiae and hemorrhage may also be present in association with the dermatitis or the papular lesions of LCH, and they may be seen both with and without associated thrombocytopenia. Other less commonly seen cutaneous presentations include nodules and granulomatous, ulcerative lesions. Box 10-2 lists some cutaneous clues to the diagnosis of LCH.
Recalcitrant seborrheic dermatitis-like eruption
Localization of rash to scalp, posterior auricular regions, perineum, axillae
Eroded papules in flexural areas
Petechial or purpuric papules
Crusted papules on palms and/or soles (scabies preparation negative)
Any of the above lesions in combination with lymphadenopathy
Mucosal involvement may occur in patients with LCH, especially those with more disseminated involvement. Gingival erythema, erosions, and hemorrhage may be seen. In some infants, gingival and oral mucosal erosions with premature eruption of teeth may be the initial manifestations of LCH. Loosening of the teeth may occur as a result of severe gingivitis, especially when there is concomitant bony involvement of the alveolar ridge and jaw. Involvement of the external auditory canals may result in chronic otitis externa.
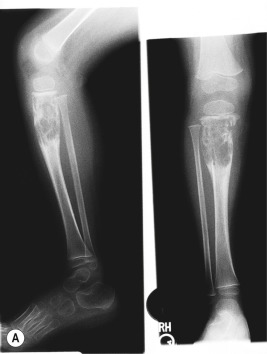
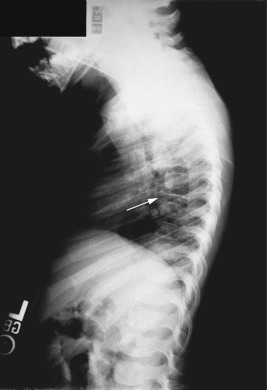
Bone lesions are very common in patients with LCH. When they occur, pain (with or without swelling) is the most common presenting complaint, and patients may complain of discomfort both during activity and when at rest. Bony involvement occurs most commonly in the skull, followed by the long bones of the extremities and the flat bones (pelvis, vertebrae, ribs). Radiographic studies usually reveal single or multiple lytic lesions of bone ( Fig. 10-6 ) that often have a “punched-out” appearance. Proptosis may result from orbital wall involvement, and the radiographic appearance may simulate mastoiditis when there is involvement of the mastoid process. Middle-ear extension may cause destructive changes with resultant deafness, and chronic otitis media may also occur as a result of mastoid and temporal bone disease. Vertebral body involvement may result in compression with the radiographic finding of vertebra plana ( Fig. 10-7 ). Plain radiographs are usually sufficient for diagnosing bony LCH, but computed tomography (CT), magnetic resonance imaging (MRI), and technetium (Tc)-99 bone scans may also be useful, especially in the setting of multifocal osseous disease.
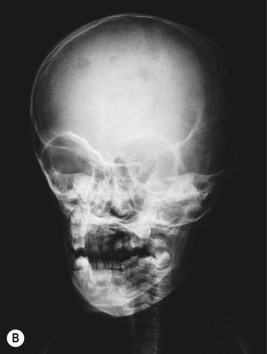
The classic triad (historically Hand–Schüller–Christian disease) of skull lesions, DI, and exophthalmos is the prototype for multifocal LCH, but many other organs may be involved in this setting. Disseminated LCH (historically Letterer–Siwe disease) is the most serious form of the disorder, differing only in extent and severity from multifocal involvement. Lymph node involvement may be seen with localized or widespread LCH, and most often the cervical nodes are involved. Hepatosplenomegaly, biliary cirrhosis, and liver dysfunction may occur. Pulmonary LCH may present with cough, hemoptysis, dyspnea, or pain and is most common in the third decade of life. Pulmonary involvement may also be asymptomatic, especially in younger children. Chest radiography reveals a diffuse micronodular or reticular pattern, and pneumothoraces may result. Gastrointestinal tract involvement may present as anorexia, malabsorption, vomiting, diarrhea, and failure to thrive. However, gastrointestinal involvement may be asymptomatic and thus is at times overlooked. Thymus abnormalities have been reported and may present with enlargement of the gland on chest radiography. Bone marrow involvement may result in pancytopenia and with concomitant hypersplenism may contribute to life-threatening sepsis and hemorrhage. Nonspecific constitutional symptoms, including fever, weight loss, and malaise, are common in patients with multiorgan involvement.
Central nervous system (CNS) involvement in LCH may include infiltration of the hypothalamic–pituitary regions, which can result in DI even years after the initial diagnosis. DI seems to develop more often in patients with bony involvement of the skull and multisystem disease. Posterior pituitary infiltration may be evident on MRI as absence of a normally bright signal in the posterior pituitary gland or thickening of the pituitary stalk. Growth retardation may result from anterior pituitary involvement, and growth hormone deficiency, which is relatively uncommon, may occur and was noted in 6% of patients in one large series. Other manifestations of CNS involvement include hyperreflexia, dysarthria, cranial nerve defects, and rarely seizures. Progressive or rarely, acute ataxia has been observed as a complication. Basilar invagination, which is associated with hydrocephalus and usually occurs as part of the Arnold–Chiari malformation or in patients with diseases that result in bone softening (i.e., osteogenesis imperfecta), has been reported in long-term survivors with LCH. MRI is useful in diagnosing CNS disease. Positron emission tomography (PET) scan has been demonstrated useful in identifying areas with altered metabolism related to CNS LCH and may provide a tool for longitudinal involvement in some patients. Neuropsychologic deficits that may occur in children with LCH include cognitive deficiencies and deficits in memory, attention and concentration, and perceptual–organizational capabilities. Cognitive defects are noted especially in patients with multisystem LCH with CNS involvement. Psychiatric deterioration and cognitive defects may both be more common in patients who develop cerebellar involvement.
Congenital self-healing reticulohistiocytosis deserves special mention. This entity, also known as Hashimoto–Pritzker disease (and also as congenital self-healing LCH [CSHLCH] ), is marked by the congenital presence of LCH lesions, usually papules and nodules, which may break down in the center and form crater-shaped ulcers. Systemic signs are often absent, and the lesions involute over a few months and are usually gone by 12 months. There may be some distinct histologic features but not always. It is generally accepted that CSHLCH is a variant of LCH and that most patients have a favorable prognosis. However, patients do not always have disease limited to the skin and thus require an evaluation for systemic involvement. In addition, reports of cutaneous and systemic relapse (including DI) months to years after resolution of the skin lesions highlight the importance of vigilant long-term follow-up observation as one would do for patients with classic LCH.
Erdheim–Chester disease is a rare, non-Langerhans systemic histiocytosis characterized by xanthomatous or xanthogranulomatous infiltration of tissues and a cellular phenotype that is CD68 positive but CD1a and S100 negative. It may present in a fashion similar to LCH, including bony involvement, although it most commonly causes osteosclerosis affecting the metaphyseal regions of long bones (and presenting with bilateral knee and ankle pain). The cutaneous lesions are best characterized as a variant of juvenile xanthogranulomas (JXGs) (see below), and the clinical findings in the skin usually consist of xanthelasma-like lesions on the eyelids. Erdheim–Chester disease occurs predominantly in adults.
LCH is diagnosed by examination of tissue specimens from affected organs. Skin is a readily accessible organ for biopsy in patients who have cutaneous involvement. Routine histologic sections reveal an infiltrate of typical Langerhans cells, which can be confirmed by positive S100, CDla, or langerin immunostaining. Electron microscopy, although rarely performed now because of the availability of special stains, reveals the characteristic Birbeck granules within the cytoplasm of the cells. The prognosis for patients with LCH is quite variable and dependent on many factors, including the extent of organ involvement. In general, the younger the age at diagnosis, the shorter the event-free survival and overall survival rates, although this association does not appear to be as strong as once believed. Interestingly, neonates with isolated cutaneous lesions tend to do very well. Other prognostic factors include the type and number of disease sites, organ dysfunction, and response to therapy. Morbidity and mortality may be related to progressive disease or late sequelae, which include skeletal defects, dental problems, DI, growth failure or other endocrinopathies, hearing loss, and CNS dysfunction. DI, which presents with polyuria and polydipsia, occurs in around 15% to 25% of patients and may even occur as a late complication in patients who present with “skin-limited” disease or CSHLCH.
The recommended evaluation for the patient suspected of having LCH is shown in Box 10-3 . Clinical stratification based extent of disease has been recommended as follows: single-organ-system disease (unifocal or multifocal), multiorgan disease (without organ dysfunction), and multiorgan disease (with organ dysfunction). The latter category, multiorgan disease with organ dysfunction, is further stratified as low risk (skin, bone, lymph node, pituitary) or high risk (lung, liver, spleen, hematopoietic cells). Patients with single-organ-system disease seem to have the best outcome and a low reactivation rate.
Physical examination, including growth parameters
Laboratory evaluation:
Complete blood cell count
Coagulation studies
Hepatic function testing
Urine osmolality
Complete skeletal radiographic survey
Chest radiography
More specific studies as guided by initial results (i.e., bone marrow examination, pulmonary function testing, lung biopsy, liver biopsy, panoramic dental films, CT or MRI of the CNS, endocrine evaluation)
CNS, Central nervous system; CT, computed tomography; MRI, magnetic resonance imaging.
Therapy for LCH depends on the extent of disease. In patients with disease limited to the skin, observation alone is often appropriate, because the lesions may resolve spontaneously. Topical corticosteroids are only occasionally effective. Topical treatment with nitrogen mustard may be used, and in patients with severe skin disease, systemic therapy as is given for multiorgan LCH may be considered. Treatment for disease limited to bone is dictated by the extent of bone involvement and symptomatology. Single-bone lesions may resolve spontaneously, and simple observation is appropriate in this setting in patients who are not experiencing pain. Painful lesions may be treated with curettage, surgical excision, or intralesional steroid injection. Localized radiation therapy is another treatment option.
In patients with more extensive LCH, there are a variety of therapeutic options. Most patients are treated with systemic chemotherapy, most commonly vinblastine or etoposide. These two agents have been found equally effective in the treatment of multisystem LCH. Prednisone or methylprednisolone is commonly used during the induction phase of therapy for patients with multisystem LCH. Other reported therapies include cyclosporine A, 2-chlorodeoxyadenosine, interferon-α, cladribine, cytarabine, and allogeneic bone marrow or cord-blood transplantation.
LCH is a potentially fatal disorder that can be recognized and diagnosed early based on the characteristic cutaneous manifestations. The skin signs may appear alone or in combination with systemic disease, and all patients require a thorough evaluation for extracutaneous involvement before they can be diagnosed as having skin-limited disease. Although there is often a delay in the diagnosis of patients with LCH, becoming familiar with the cutaneous clues (see Box 10-2 ) will optimally position the pediatrician or other pediatric healthcare providers to recognize the disorder. Patients with LCH are usually treated by a pediatric oncologist, and all patients require long-term surveillance for late sequelae or relapse.
Juvenile Xanthogranuloma
JXG is a common form of non-LCH. It is generally a benign, self-limited disease of infants, children, and occasionally adults. Lesions occur most often in skin, although extracutaneous disease may occasionally be present. Most JXGs occur early in life, and the true incidence may be underestimated because many of them may go undiagnosed or misdiagnosed as other common skin tumors such as nevi. JXG seems to be derived from dermal dendrocytes, and although the term xantho- appears in the name, there is no association of this condition with hyperlipidemia or other metabolic abnormalities.
JXG presents as a firm, round papule or nodule, varying in size from 5 mm to 2 cm, with giant lesions (i.e., up to 5 or 10 cm) occasionally seen. Some authors have divided JXG into a “micronodular” form (lesions <10 mm) and a “macronodular” form (lesions >10 mm). Early JXGs are erythematous ( Fig. 10-8 ) to orange or tan ( Fig. 10-9 ), but with time they become more yellow in color ( Figs. 10-10 and 10-11 ). Lesions may be solitary (up to 90% of all patients with JXG ) or less commonly multiple ( Fig. 10-12 ), and they are usually asymptomatic. Ulceration and crusting may occasionally occur. The head, neck, and trunk are the most common areas to be involved. Lesions may also occur on mucous membranes or at mucocutaneous junctions (mouth, vaginal orifice, and perineal area). Oral lesions occur on the lateral aspects of the tongue, gingival, buccal mucosa, and midline hard palate and may ulcerate and bleed. Oral lesions may appear verrucous, pedunculated, umbilicated, or fibroma-like. Typically, the lesions of JXG present at birth (20%) or during the first 6 months of life, and they may persist or continue to erupt for years.
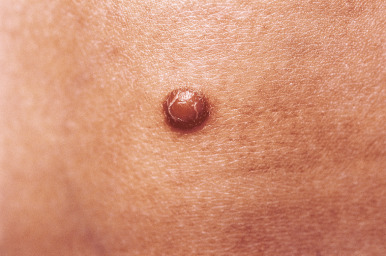
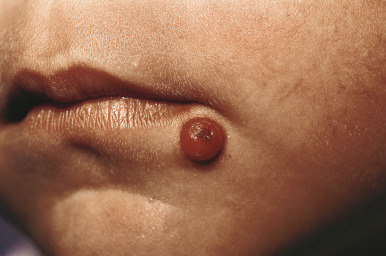
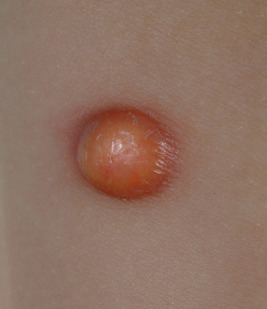
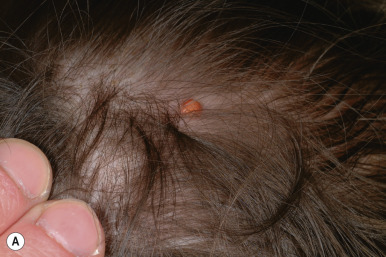
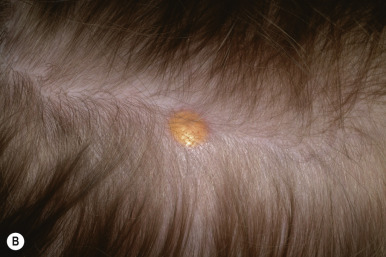
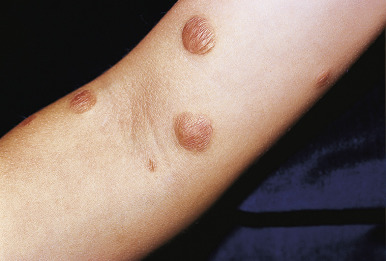
Histologic evaluation of JXG tissue reveals a dense dermal infiltrate of foamy histiocytes, foreign body cells, and the characteristic Touton giant cells, which are virtually pathognomonic for the condition. The Touton giant cell is a giant cell with a central wreath of nuclei and a peripheral rim of eosinophilic cytoplasm. Lymphocytes and eosinophils are often seen, and the histiocytes in JXG are S100 and CDla negative on special staining.
Extracutaneous involvement occasionally occurs with JXG. The eye is the most common organ of involvement second to the skin. The iris is the site most often involved, and potential complications of ocular JXG include hyphema, glaucoma, or blindness. Patients may complain of eye redness, irritation, or photophobia. Children at greatest risk for ocular JXG include those 2 years of age or younger and those with multiple skin lesions. Intramuscular JXG presents as a deep, soft-tissue lesion that may have imaging features similar to those of malignant tumors of infancy. This form tends to affect exclusively infants and toddlers and occurs as a solitary lesion in skeletal muscles of the trunk. Other sites of extracutaneous involvement include lung, liver, testis, pericardium, spleen, CNS, bone, kidney, adrenal glands, and larynx. Solitary as well as multiple intracranial and intracerebral lesions have rarely been reported, including one patient who experienced leptomeningeal spread and several who required therapy with chemotherapy and steroids. Intraspinal JXG with spinal-cord compression has been reported. Systemic JXG generally exhibits a benign clinical course but may occasionally be fatal, especially when the liver is involved. There are rare reports of JXG in association with LCH, suggesting a possible common progenitor cell and overlap within the histiocytic spectrum of disorders.
An important association is that of JXG and childhood leukemia. The most common association has been with juvenile chronic myelogenous leukemia (JCML), which may be seen with increased incidence in patients with multiple JXG lesions. It has been noted that several such reported patients also had café-au-lait macules and a family history of type 1 neurofibromatosis (NF). A systematic review of the literature revealed that the frequency of the triple association of JXG, JCML, and NF is 30- to 40-fold higher than expected, and it is estimated that children with NF and JXG have a 20- to 32-fold higher risk for JCML than do patients with NF who do not have JXG. However, it should be noted that the vast majority of patients with multiple JXG, even those with associated NF, do not develop JCML. The role of surveillance complete blood cell counts is controversial, and most practitioners monitor these patients primarily with regular, thorough physical examinations.
JXG usually runs a fairly benign course, with spontaneous regression occurring over 3 to 6 years. Pigmentary alteration, atrophy, or “anetoderma-like” changes may persist in areas of prior skin involvement. Although rare cases lasting until adulthood have been reported, generally those that have their onset early in life manifest complete spontaneous healing. The risk of complications is fairly high when ocular involvement is present. For this reason, once disease has been confirmed in the eye, therapy should be initiated. Intraocular JXG is treated with intralesional or systemic steroids, radiation therapy, or excision. Lesions limited to skin require no therapy, although surgical excision is occasionally performed for diagnostic or cosmetic purposes. Systemic involvement is treated if it interferes with vital functions and has shown response to chemotherapy regimens similar to those used in LCH. Patients with JXG and NF should be monitored for the development of leukemia given the increased risk.
Xanthoma Disseminatum
Xanthoma disseminatum (XD), a rare disorder of mucocutaneous xanthomatous lesions, is another non-LCH. This disorder usually occurs in adults, although it may have its onset during childhood. Patients have numerous (sometimes hundreds) round to oval, yellow-orange or brown papules, nodules, and plaques. They occur primarily on the face and the flexural and intertriginous surfaces, including the neck, antecubital fossae, periumbilical area, perineum, and genitalia. The lips, eyelids, and conjunctivae may be involved, and xanthomatous deposits have also been observed in the mouth and upper respiratory tract (epiglottis, larynx, and trachea), occasionally leading to respiratory difficulty. Facial lesions may become exuberant and may cause disfiguration. Osseous lesions, presenting radiographically as well-demarcated areas of osteolysis, may be present. Ocular mucosal lesions may result in blindness. Liver involvement is occasionally present. When XD involves the CNS, it is characterized by infiltration of the pituitary gland and termed xanthomatous hypophysitis, and may result in DI, hyperprolactinemia, or hypopituitarism.
As with JXG, there is no perturbation in lipid metabolism in patients with XD, although it has been rarely reported in affected children. DI occurs in many patients with the disorder, and severe laryngeal involvement may necessitate tracheostomy. The lesions of XD often persist indefinitely but have been known to involute spontaneously. Treatment of the cutaneous lesions has been performed with cryotherapy, excision, and carbon dioxide laser ablation. Despite the normolipemic nature of XD, lipid-lowering agents have been utilized in some patients with reported improvement. Respiratory tract involvement, when severe, may justify a more aggressive approach with localized radiation therapy or chemotherapy.
Benign Cephalic Histiocytosis
Benign cephalic histiocytosis (BCH) is a self-healing, cutaneous, non-LCH that classically involves the face and head. The average age of onset is 15 months, and 45% of cases occur in infants under 6 months of age. Clinically BCH is characterized by small, 2- to 6-mm, yellow-brown macules and minimally elevated papules ( Fig. 10-13 ). The lesions may occasionally coalesce to give a reticulate pattern. BCH most commonly occurs on the face and head (more than 80% of 45 patients in one review) and less commonly on the neck and trunk. The extremities, buttocks, and pubic area may be involved later in the course. The differential diagnosis may include flat warts, micronodular JXG, LCH, multiple melanocytic nevi, and urticaria pigmentosa.
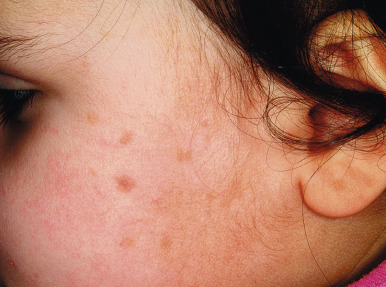
The diagnosis of BCH can be confirmed by skin biopsy, which reveals a histiocytic infiltrate with negative stains for S100 and CDla. Electron microscopy is useful in confirming the diagnosis, because the cells classically reveal intracytoplasmic comma-shaped or worm-like bodies and the absence of Birbeck granules, clearly differentiating it from LCH.
There is significant clinical overlap between some of the non-LCHs. Some authors have considered BCH to be a variant of generalized eruptive histiocytoma (GEH) (see below). In addition, there are reports of BCH progressing into JXG, and although BCH tends to be limited to the skin, there are some reports of associated internal involvement. One patient with BCH developed DI 1 year later with infiltration of the pituitary stalk on imaging. In another patient with classic lesions of BCH on the face, lytic lesions in the skull, spine, and tibia were demonstrated to be LCH on tissue examination. These clinical observations again highlight the potential overlap among the histiocytic syndromes.
BCH generally runs a benign course with spontaneous healing, often leaving behind flat or atrophic pigmented scars. Treatment is unnecessary, but given the clinical variation in presentation and overlap with other histiocytic syndromes, clinical follow-up observation for progression or internal involvement is advisable.
Necrobiotic Xanthogranuloma
Necrobiotic xanthogranuloma, a rare disorder usually reported in adults, is characterized by sharply demarcated, indurated plaques and nodules that are usually yellow to red-brown or violaceous and have a predilection for periorbital areas, the trunk, and proximal extremities. Lesions vary in size, may at times be as large as 10 cm or more in diameter, and often ulcerate and heal with areas of atrophy and telangiectasia. Ocular involvement is fairly common and includes ectropion, ptosis, keratitis, uveitis, proptosis, and orbital masses. Visceral lesions of necrobiotic xanthogranuloma may involve the heart, skeletal muscles, larynx, spleen, and ovary. A hallmark of this disease is an associated paraproteinemia, immunoglobulin (Ig) G monoclonal gammopathy. The course of necrobiotic xanthogranuloma is chronic and progressive and may be associated with proliferation of plasma cells in the bone marrow or multiple myeloma. Treatment options include systemic or intralesional corticosteroids, cytotoxic drugs, radiation therapy, and plasmapheresis. Intravenous immunoglobulin has been reportedly useful in some patients. Surgical removal of lesions is associated with a high recurrence rate.
Generalized Eruptive Histiocytoma
Generalized eruptive histiocytoma (GEH) is a rare, self-healing histiocytosis characterized by recurrent crops of small, yellow to red-brown papules on the face, trunk, extensor extremities, and rarely, mucous membranes. The lesions number in the hundreds and tend to occur in a symmetric distribution. They resolve over months with hyperpigmented macules left in their place. As a result of the continuous development of new lesions, however, the disorder may persist indefinitely. Although generally regarded as a disorder of adults, GEH has been seen in childhood and even in infants as young as 1 month of age.
The differential diagnosis includes JXG (which may be very difficult to differentiate histologically from early lesions of GEH), LCH, BCH, and urticaria pigmentosa. XD has developed in a child with the prior diagnosis of GEH. No treatment is generally necessary for this disorder. However, since there is considerable overlap within non-LCH, patients should be monitored carefully for the development of signs or symptoms of other organ involvement. Isotretinoin treatment has been reported in a patient with GEH, with transient improvement but eventual recurrence.
Progressive Nodular Histiocytoma
Progressive nodular histiocytoma is characterized by a widespread eruption of hundreds of yellow-brown, 2- to 10-mm papules and deeper, larger subcutaneous nodules. Conjunctival, oral, and laryngeal lesions may occur. The face typically is heavily involved with numerous coalescent lesions, which may result in a leonine appearance. New lesions progressively occur, and ulceration is common. Occasionally, bleeding may occur within the subcutaneous nodules, resulting in marked pain. Lesions of progressive nodular histiocytoma histologically show features typical of xanthogranuloma, and some authors suggest that this entity and JXG may represent a variation of the same process. Treatment for this condition is difficult and includes excision of large or symptomatic lesions, chemotherapy with vinblastine, and electron beam therapy.
Multicentric Reticulohistiocytosis
Multicentric reticulohistiocytosis is a rare systemic disorder of unknown cause. It is characterized by cutaneous lesions and a destructive arthritis and is seen almost exclusively in adults (mean age at diagnosis of 40 to 50 years) with rare reports of pediatric disease. The skin lesions present as firm red-brown papules and nodules, most often distributed on the hands, fingers, lips, ears, and nose. Facial disfigurement may occur, with cartilaginous destruction and the appearance of a leonine facies. The coral bead sign refers to the presence of a chain of papules along the cuticle. Nodular lesions on the arms, elbows, and knees may occur and at times resemble rheumatoid nodules. Mucous membrane involvement may occur in up to one-half of patients. They most often present on the lips, buccal mucosa, nasal septum, tongue, palate, and gingivae.
Joint involvement is the presenting sign in more than half of the patients and is highly destructive. It may involve any joint, especially the interphalangeal joints, and coexisting synovitis is common. Shortening of the digits may occur with a “telescopic” or “opera-glass” deformity. Rheumatoid arthritis may be mistakenly diagnosed in some patients. Microscopic examination of skin, bone, or synovial tissue reveals a characteristic histiocytic process with multinucleated giant cells and a “ground-glass” appearance. Visceral involvement may include pleural effusion, pulmonary fibrosis, pericardial effusion, congestive heart failure, salivary gland enlargement, muscle weakness, lymphadenopathy, and gastric ulcer. Multicentric reticulohistiocytosis may regress spontaneously over 6 to 8 years, but for many patients the articular destruction results in permanent joint deformities. The response of this disorder to therapy is often disappointing, with treatments including nonsteroidal anti-inflammatory agents, corticosteroids, cyclophosphamide, chlorambucil, methotrexate, hydroxychloroquine, infliximab, tocilizumab, and interferon.
Hemophagocytic Lymphohistiocytosis
Hemophagocytic lymphohistiocytosis ( HLH ; also known as hemophagocytic syndrome ) refers to a condition characterized by fever, wasting, jaundice, and hepatosplenomegaly resulting from diffuse infiltration of phagocytizing histiocytes in various tissues. This disorder is heterogeneous in its etiologies, which include a familial form (familial HLH) and a secondary/reactive form that may be associated with a variety of infectious agents (most notably viral, bacterial, or parasitic), malignancy, and collagen vascular disorders. These have also been referred to as genetic and acquired forms of HLH. The infection-associated form is seen primarily in immunocompromised patients with evidence of preceding viral (usually Epstein–Barr virus [EBV]) infection, in which case it is also known as virus-associated hemophagocytic syndrome. The other viral agents reported in association with HLH include cytomegalovirus, enterovirus, and parainfluenza virus. The term malignant histiocytosis has also been used to describe this condition, in response to the cytologic atypia and malignant nature of the infiltrating histiocytes.
HLH has been observed as a complication of allogeneic hematopoietic stem-cell transplantation and in association with hematologic malignancies. There also seems to be a relationship between EBV-associated HLH and EBV-associated T-cell lymphoma, with HLH representing the major cause of death in these patients. Lymphoma-associated hemophagocytic syndrome (LAHS) is a rarely reported variant of HLH that has been described in the setting of systemic and cutaneous lymphomas, including subcutaneous panniculitic T-cell lymphoma (SPTCL) and epidermotropic cutaneous T-cell lymphoma, in which case it often occurs several years after the lymphoma diagnosis. The pathway most commonly implicated in familial HLH is that of perforin-dependent cytotoxicity, an essential function of natural killer and cytotoxic T lymphocytes. The genetic forms of HLH have been shown to be related to mutations in any of several genes involved in this pathway, including PRF1, UNC13D, Munc18-2, Rab27a, STX11, SH2D1A, and BIRC4 . HLH may also be related to a primary immunodeficiency syndrome. Macrophage activation syndrome (see Chapter 22 ) is a severe condition with features very similar to HLH that is seen in association with the systemic form of juvenile idiopathic arthritis and, less often, other autoimmune inflammatory diseases. Box 10-4 summarizes the various subtypes of HLH.
Genetic Forms
Familial hemophagocytic lymphohistiocytosis (FHL)
FHL1–FHL5
Immunodeficiency disorders
Griscelli syndrome type 2
Chediak–Higashi syndrome
Hermansky–Pudlak syndrome type 2
X-linked proliferative syndrome (XLP), types 1 and 2
IL-2 inducible T-cell kinase (ITK) deficiency
Acquired Forms
Viral (especially EBV and other herpes viruses)
Other infections (bacterial, fungal, parasitic)
Malignancy (peripheral lymphomas, cutaneous lymphomas, leukemia, myeloma, some solid tumors)
Immunosuppression (post-organ transplantation or post-HSCT)
Autoimmune disease (systemic-onset JIA, SLE, spondyloarthropathies, Kawasaki disease; termed macrophage activation syndrome )
EBV, Epstein-Barr virus; HSCT, hematopoietic stem cell transplantation; JIA, juvenile idiopathic arthritis; SLE, systemic lupus erythematosus.
The cutaneous manifestations seen in patients with HLH are variable. Most common is a transient, generalized maculopapular eruption. Petechial and purpuric macules, generalized erythroderma, and morbilliform erythema may also occur. Although the skin findings are not specific, their presence in the patient with a supportive history and/or the concomitant findings of fever, lymphadenopathy, hepatosplenomegaly, and cytopenias should prompt consideration for this diagnosis. The most common hematologic findings are leukopenia and thrombocytopenia, and coagulopathy is fairly common. Other findings may include liver dysfunction and elevated triglyceride, lactate dehydrogenase (LDH), and ferritin levels. The clinical differential diagnosis may include extramedullary hematopoiesis (as may be seen with a variety of infectious or malignant disorders) and metastatic lesions from an underlying malignancy. LCH and myofibromatosis may also be in the differential. Skin biopsy may be useful in eliminating some diagnoses, but the histologic findings are often nonspecific and the changes of erythrophagocytosis are often absent in skin specimens. Examination of bone marrow or other solid organ (lymph node, spleen, liver) biopsy tissue may be necessary to confirm the diagnosis. HLH has a high mortality rate, although chemotherapy, steroids, intravenous immunoglobulin, and hematopoietic stem-cell transplantation may offer hope for some patients. Etoposide and dexamethasone often induce clinical remission of the inflammatory symptoms, and improved survival has been noted with newer reduced-intensity transplant conditioning protocols. Cyclosporine A has been utilized to inhibit T-cell activation.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


