Because of the increasing public awareness and incidence of skin cancer, physicians are often consulted regarding tumors of the skin. In children, the vast majority of cutaneous tumors are benign, and their importance lies predominantly in the cosmetic defect they may create or in their occasional association with systemic disease. Malignant skin lesions, however, despite their relative rarity in children, cannot be completely disregarded or ignored. Each lesion in children, as in adults, must be assessed individually with a consideration of its cosmetic effect, its possible association with systemic manifestations, and its capacity for malignant degeneration.
Cutaneous tumors can be differentiated into those arising from epidermal (or mucosal) cells, from melanocytes, from the epidermal appendages, or from dermal or subcutaneous cells or tissues. The latter category includes tumors of fibrous, neural, vascular, fatty, muscular, and osseous tissues. Skin tumors can also be divided into benign (the vast majority of lesions in children) and malignant. The term nevus (plural nevi ) has a broad meaning in dermatology. Strictly defined, this term refers to a circumscribed congenital abnormality of the skin. When this term is used, therefore, it is appropriate to include a qualifying adjective (i.e., epidermal nevus, melanocytic nevus, or vascular nevus), thus specifying the cell of origin. In commonplace practice, however, the term nevus is usually used to imply a benign tumor of pigment cells (melanocytes). Hence to many practitioners, a nevus is the same as a “mole.”
In this chapter, we will discuss several cutaneous tumors (and corresponding tumor syndromes where applicable). Pigmented lesions are discussed as a separate entity apart from the above classification, given their high incidence.
Pigmented Lesions
Pigmented lesions, especially melanocytic nevi (moles), are the most common neoplasms found in humans. In this section, we will discuss melanocytic nevi (both congenital and acquired) and several variants, including dysplastic, Spitz, and halo types. Nevus spilus and Becker nevus, two distinct pigmented lesions, will also be discussed, as will malignant melanoma (MM), which although rare in childhood, does occasionally occur.
Melanocytic Nevi
Melanocytic nevi are extremely common skin neoplasms composed of “nevocytes” or nevus cells that are believed to be slightly altered melanocytes. The melanocyte is a dendritic cell that produces melanin and transfers it to keratinocytes (epidermal cells) and hair cells, thus supplying the normal brown pigment to skin and hair. Both melanocytes and nevus cells are of neural origin. Melanocytes originate in the neural crest and early in fetal life migrate from there to the skin. After birth some melanocytes occasionally remain in the dermis of certain races (Asians, Native Americans, African-Americans, and individuals from the Mediterranean region), where they may appear as Mongolian spots (see Chapter 11 ). Blue nevi and the nevi of Ota and Ito (see Chapter 11 ) also represent examples of arrested melanocytic migration in which the melanoblasts remain in the dermis.
Melanocytic nevi can be either congenital (described in more detail below) or acquired. Acquired nevi usually appear after infancy, increase in size and number during early childhood, and peak during the third or fourth decade, with slow involution as aging progresses. The predisposition of an individual to the development of acquired melanocytic nevi seems to be related to several factors, including skin type, race, genetic predisposition, and ultraviolet light exposure. These lesions tend to be few in childhood, increasing in number with age to a peak in the third decade. Sun exposure, sunburns, and fair skin pigmentation seem to be associated with their development in childhood. In one large cross-sectional study, nevus counts were found to steadily increase with age from a median of three at age 2 years to 19 at age 7 years. High numbers of nevi were associated with moderate sun exposure and outdoor activities. In another study, 5- and 6-year-old children with a history of sunburns or an increased number of holidays in foreign countries with a sunny climate had significantly higher nevus counts than controls without these characteristics. Higher numbers of childhood nevi have been demonstrated in areas of skin chronically exposed to the sun and in children with lighter skin, blond hair, and blue eyes. Higher nevus counts are noted in children who reside in sunny climates (i.e., Australia) when compared with age- and race-matched control children. Melanocytic nevi may also occur in higher numbers in children with a history of leukemia and/or a history of chemotherapy, and in these settings the lesions may occur with greater incidence on acral areas such as the palms and soles.
The primary importance of melanocytic nevi lies in their possible transformation into MM. These lesions are felt to be both markers of an increased risk of cutaneous melanoma and in some cases, direct precursor lesions. The annual transformation rate of a single mole into melanoma seems extremely low, with an estimate of 0.0005% or lower for individuals younger than 40 years. Nonetheless, melanocytic nevus cells were seen histologically in proximity to MM in 51% of cases in one study. Genetic analyses of nevi and melanoma reveal nonrandom patterns of genetic alteration (loss of heterozygosity), confirming the notion that the former are precursor lesions for the latter. These findings highlight the relevance of close monitoring of melanocytic nevi for atypical features as well as the importance of sun protection education.
Melanocytic nevi are subdivided into types and clinically described based on the microscopic location of the nevus cells. Accordingly they are classified as junctional, intradermal, or compound lesions. Junctional nevi have proliferation of nevocytes at the dermal–epidermal junction, compound nevi reveal cells at the junction and in the dermis, and intradermal nevi reveal loss of the epidermal component with nests of cells limited to the dermis. Interestingly, these histologic subtypes progress in parallel to the natural history of nevus maturation (junctional nevi early in life, then compound nevi, and finally intradermal nevi with eventual atrophy of the dermal component during later adulthood). Melanocytic nevi have a wide range of clinical appearances. They may occur anywhere on the cutaneous or mucocutaneous surface and may be flat, slightly elevated, dome-shaped, nodular, verrucous, polypoid, cerebriform, or papillomatous.
Junctional Nevi
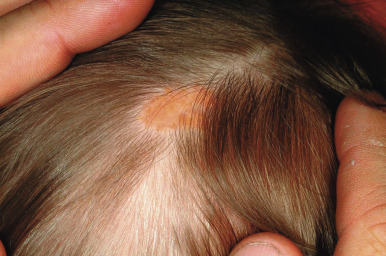
Junctional nevi usually occur as hairless, light to dark brown or black macules ( Fig. 9-1 ). They range from 1 mm to 1 cm in diameter, with a smooth and flat (nonpalpable) surface and preservation of skin furrows. Although most junctional nevi are round, elliptical, or oval and show a relatively uniform pigmentation, some may be slightly irregular in configuration and color. Most junctional nevi represent a transient phase in the development of compound nevi and are found only in children. An exception to this rule, however, is seen on the palms, soles, and genitalia where the lesions often retain their junctional appearance.
Compound Nevi
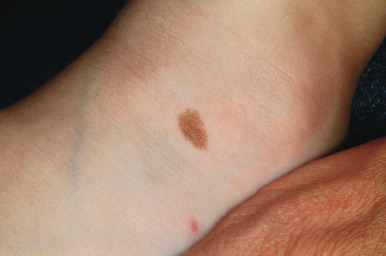
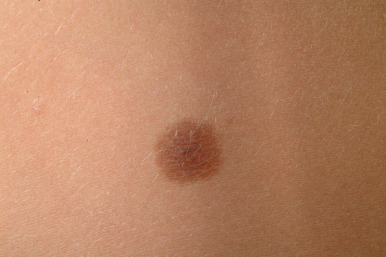
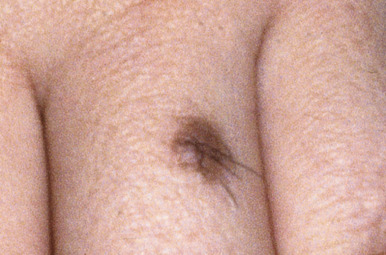
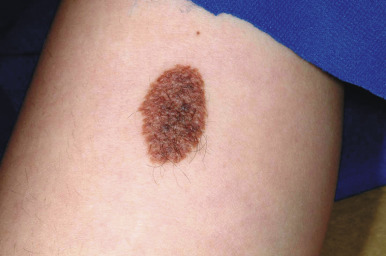
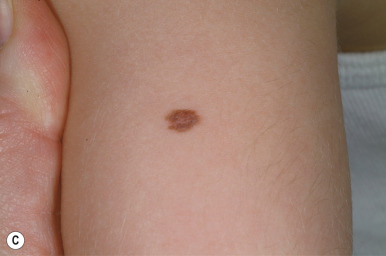
Compound nevi are more common in older children and adults but may also be present in younger children. They may appear similar to junctional nevi but tend to be more elevated and accordingly vary from a slightly raised papule to a larger, papillomatous papule or plaque. They are flesh-colored to brown ( Fig. 9-2 ), may have a smooth or verrucous surface, and may have dark coarse hairs within them ( Fig. 9-3 ), especially when occurring on the face. During later childhood and adolescence, compound nevi tend to increase in thickness and depth of pigmentation. It is at this stage that many children are brought to the physician for evaluation.
Intradermal Nevi
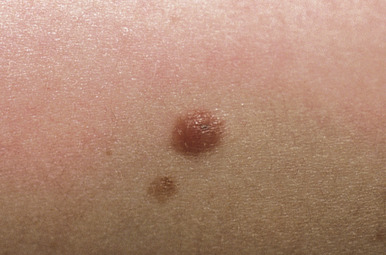
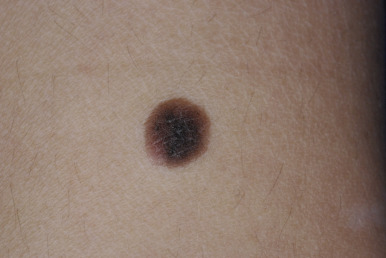
Intradermal nevi are seen most commonly in adults. They are usually dome-shaped, soft, “fleshy” papules ( Fig. 9-4 ). They may be sessile (attached by a broad base) or more pedunculated (attached by a more narrow base) and range from a few millimeters to 1 cm or more in diameter. Intradermal nevi may be clinically indistinguishable from compound nevi, and their color varies from nonpigmented (flesh-colored) lesions to those of varying shades of brown. They may occur anywhere on the skin surface and are often found on the head and neck; coarse hairs often are present. After the third decade of life, as nevus maturation continues, there is destruction and replacement of nevus cells by fibrous or fatty tissue, and by 70 years of age most individuals have few remaining nevi. In fact, nevi that persist into old age appear to have an increased risk of malignant degeneration.
Treatment decisions regarding melanocytic nevi are usually related to their cosmetic appearance, repeated irritation of the lesion, or fear of potential malignant transformation. The majority of melanocytic lesions require no treatment; by careful clinical evaluation the patient can often be reassured as to their benign nature. Removal of nevi, when indicated, is best achieved by punch biopsy or complete surgical excision. Any nevus being removed because of concern for malignant degeneration should be removed with a full-depth excision, because microscopic tumor depth is the primary prognostic indicator in MM. Every excised nevus should be subjected to histopathologic examination.
Many authors advocate for the routine excision of pigmented lesions in certain anatomic locations (i.e., palms, soles, scalp, and genitalia), owing to the belief that the likelihood of malignant transformation is greater in these areas. It seems that prophylactic excision of all nevi in these locations is unwarranted. However, lesions in acral sites may reveal atypical clinical and histologic features, possibly in relation to repetitive trauma. Recognition of these histologic variations is vital in order to avoid the misdiagnosis of the “acral lentiginous” subtype of MM, which occurs primarily after the seventh decade of life. Additionally, scalp nevi in children (and particularly adolescents) may commonly reveal mild clinical atypia, both clinically and histologically, although the vast majority behave in a biologically benign fashion (see more below). Scalp nevi that reveal significant atypia and/or that may be difficult to monitor clinically, however, may merit excision.
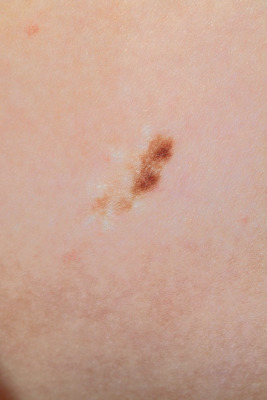
Occasionally, pigmented nevi show recurrence after excision, which may be a significant source of anxiety for the patient, parent, and at times the physician. Recurrent lesions (which have been called pseudomelanoma by some) present as circumscribed pigmentation within the surgical scar ( Fig. 9-5 ). This finding is usually not an indication of malignancy but instead represents proliferation of nevus cells from the peripheral epidermis, sweat ducts, or hair follicles. Options for management include close clinical follow-up care (assuming the original pathologic examination revealed no atypical findings) or repeat excision.
Meyerson Nevus
The term Meyerson nevus (also known as Meyerson phenomenon or halo dermatitis ) refers to a localized eczematous eruption that occurs in the region of a melanocytic nevus and was originally described by Meyerson in 1971. The inflammatory reaction does not appear to be a marker for atypia within the nevus, although there are reports of the phenomenon in association with dysplastic lesions. The Meyerson phenomenon may be associated with acquired or congenital melanocytic nevi (CMN). It has also been described in association with nonmelanocytic lesions including nevus sebaceous, dermatofibroma, and smooth muscle hamartoma. The Meyerson nevus presents clinically as a scaly erythematous plaque overlying a pigmented nevus. The overlying dermatitis may be treated with a topical corticosteroid, which often leads to complete resolution, enabling closer inspection of the underlying nevus ( Fig. 9-6 ). If left untreated, the dermatitis often resolves spontaneously over time.
Melanonychia Striata
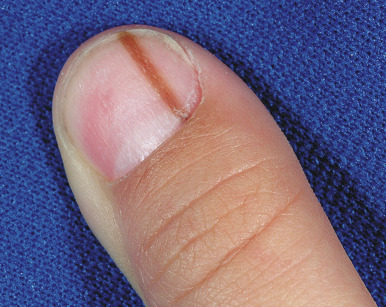
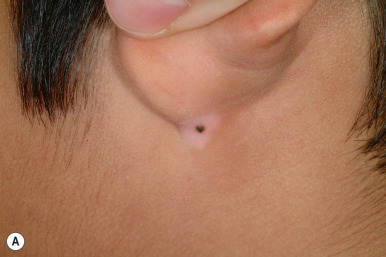
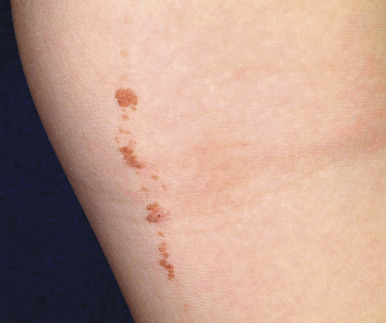
Melanonychia striata (also known as longitudinal melanonychia ) is most commonly seen in individuals with darker skin complexions, especially African-Americans, of whom up to 90% may have at least one such streak. It is significantly less common in children with white skin. Melanonychia striata presents as a brown to brown/black linear band of pigmentation on a fingernail ( Fig. 9-7 ) or toenail. The pigmentation extends from the proximal nailfold to the distal margin of the digit, and the width may vary from less than 1 mm to several millimeters. Drugs, pregnancy, trauma, and human immunodeficiency virus (HIV) infection may all be associated with melanonychia striata, although the majority of patients have no underlying association.
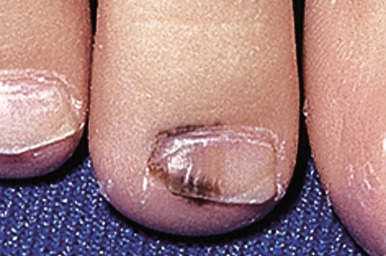
Melanonychia striata may represent benign melanocytic hyperplasia (i.e., melanocytic nevus or lentigo) or a nail matrix melanoma. In children, the majority of cases seem to be related to benign lesions, although the possibility of melanoma must always be entertained. The presence of multiple bands, as occasionally seen in darker races, is reassuring and mitigates against malignancy. Worrisome features may include very dark, broad bands and extension of the pigmentation onto the proximal or lateral nailfolds (Hutchinson sign; Fig. 9-8 ). Dermatoscopic evaluation may be useful for monitoring in the hands of clinicians experienced in its use, but the patterns and accuracy remain to be confirmed in this setting. In patients with melanonychia striata and any such atypical or concerning features, nail matrix biopsy for histologic evaluation should be highly considered. Although this procedure may result in permanent nail plate dystrophy, this risk is justified when concerns for possible melanoma exist.
Congenital Melanocytic Nevi
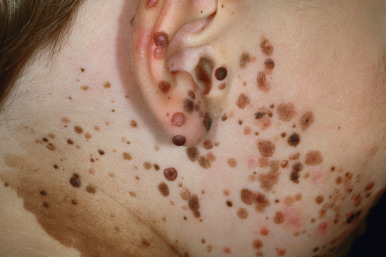
CMN represent a special group of melanocytic lesions with an increased risk of transformation to MM. By strict definition, these lesions are present at birth. However, in some patients with small and medium-sized lesions, they may be initially noted sometime during the first year of life rather than immediately at birth. Congenital nevi have been classified traditionally according to their greatest diameter in adulthood. According to this nomenclature, they have been traditionally defined as small (<1.5 cm in greatest diameter), medium-sized (1.5 to 19.9 cm), or large (≥20 cm) congenital nevi. An interdisciplinary group of experts in the field developed a consensus-based categorization scheme for CMN that includes modified size categories (again, based on projected adult size) as follows: small (<1.5 cm), medium (M1: 1.5 to 10 cm; M2: >10 to 20 cm), large (L1: >20 to 30 cm; L2: >30 to 40 cm), and giant (G1: >40 to 60 cm; G2: >60 cm). This classification includes other descriptors of CMN, including number of satellite nevi, anatomic localization, color heterogeneity, surface rugosity, hypertrichosis, and presence of dermal or subcutaneous nodules. Such a standardized classification scheme will hopefully facilitate collaborative international studies and database development for the study of large and giant CMN.
It is estimated that approximately 1% of newborns have a small congenital nevus, and large lesions occur in around 1 in 20,000 newborns. Giant melanocytic nevi occur in 1 in 500,000 newborns. Nevus spilus, or speckled lentiginous nevus (see below), may represent a distinct subtype of congenital melanocytic nevus that presents early in life as a café-au-lait macule, with the development of the more characteristic nevi a bit later in life. Whereas malignant potential may be a primary concern with CMN, the potential psychosocial consequences, especially with large or giant lesions, are significant and may include social difficulties, psychologic morbidities, behavioral and emotional impairment, and predisposition to taunting or bullying.
Congenital nevi have some characteristic histopathologic features, including extension of nevus cells into the deeper dermis and subcutis, between the collagen bundles (as single cells), and in association with appendages, nerves, and vessels. These features may be useful when histologic confirmation of a congenital lesion is desired, but such findings may not be noted in every congenital nevus. This pattern of deep extension is important to keep in mind when surgical intervention of such a lesion is being considered. Activating mutations in the NRAS gene have been noted within congenital nevi, distinct from nevi developing after birth (acquired nevi), which are more likely to reveal mutations in BRAF (see also below). In a series of large and giant CMN, NRAS mutations were noted in nearly 95% of cases, whereas in small- and medium-sized CMN, 70% bore NRAS mutations and 30% revealed BRAF mutations. In a series of patients with multiple CMN and neurocutaneous melanosis (see below), both the cutaneous and neural lesions contained missense mutations in codon 61 of NRAS , whereas unaffected tissues and blood were negative for this mutation, suggesting postzygotic mutation in a progenitor cell within the neuroectoderm.
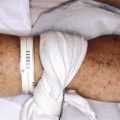
Small and medium-sized CMN usually present as flat, light- to dark-brown or pink macules ( Fig. 9-9 ) or papules or as brown to dark brown, well-circumscribed patches or plaques. They may reveal some pigment variation and the surface may have hair ( Fig. 9-10 ). With time they tend to become more elevated, and coarse dark brown hairs may become more prominent. When an extremity is circumferentially involved with a medium- or large-sized congenital nevus, limb underdevelopment may occur ( Fig. 9-11 ).
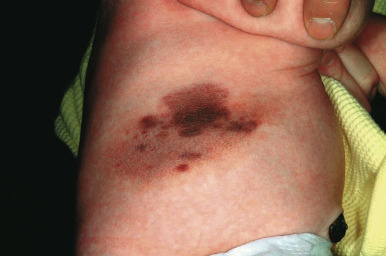
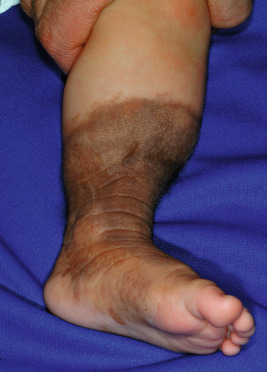
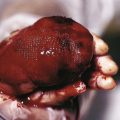
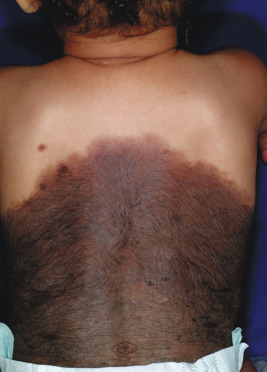
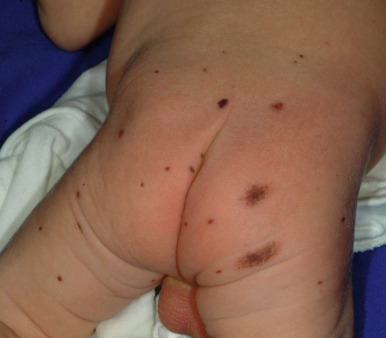
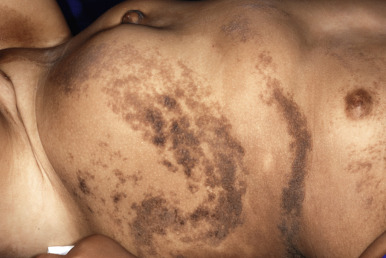
Large CMN are uniformly present at birth and present as dark brown to black plaques, often with a verrucous or cobblestoned surface and hypertrichosis ( Fig. 9-12 ). Color variation is common in these lesions and may make clinical examination for concerning features more challenging. Giant CMN often occur on the posterior trunk and may occupy a significant portion of the skin surface. They may have a dermatomal distribution and may involve an entire upper or lower extremity or the scalp. These lesions have been variably named coat-sleeve, stocking, bathing trunk, or giant hairy nevi, depending on their site(s) of involvement. As with large CMN, giant nevi present as large, brown to black plaques with varying degrees of nodularity, color variation, and hypertrichosis ( Fig. 9-13 ). Erosions or ulcerations in these giant lesions may occur ( Fig. 9-14 ), and although once believed to be consistently ominous findings, are often benign and usually not indicative of MM. Large and giant congenital nevi may be associated with cosmetic disfigurement, an increased risk of MM, and underlying neurocutaneous melanosis (see below). These lesions often have associated “satellite” nevi that may be disseminated over the entire skin surface. Satellite nevi are usually tan to brown macules or papules, with varying degrees of hypertrichosis ( Fig. 9-15 ). They may be present at birth or continue to develop during infancy.
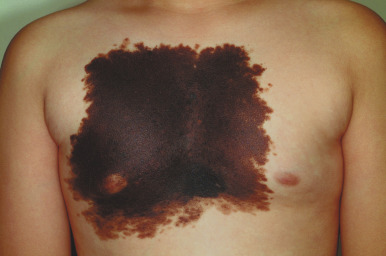
The congenital nature of nevi is one of several known risk factors for MM, but the exact magnitude of risk remains controversial, especially for small and medium-sized lesions. All congenital nevi should be considered as potential precursors to melanoma. The risk in small and medium-sized lesions seems low, and if melanoma occurs it tends to occur during adulthood. Factors that might increase the level of concern include atypical clinical features (e.g., deeply or irregularly pigmented or rapidly growing), an abnormal nevus phenotype, or a strong family history of MM. The risk of malignant transformation of congenital nevi in African-American patients is extremely small.
The risk of MM in large and giant CMN appears to be significantly greater than that for small and medium-sized lesions, with melanoma often (but not always) occurring before the age of 5 years. It is unclear whether axial lesions pose a greater risk than those occurring on the extremities, as traditionally suggested. When melanoma occurs in patients with large or giant CMN, it may occur within the skin or in extracutaneous sites such as the central nervous system. Development of melanoma in satellite lesions is exceedingly unlikely. The overall lifetime risk of development of MM in patients with large CMN has been reported to range from 0% to 31% but is best estimated at between 2% and 10%. In two large systematic reviews of the published data on melanoma risk in CMN, the rate of melanoma was 2% to 2.5% in large CMN and 3.1% in giant CMN. In addition to melanoma, other malignancies may occur with increased rates in these patients, including rhabdomyosarcoma, liposarcoma, malignant peripheral nerve sheath tumor, and other sarcomas.
Large and giant CMN, particularly those on the head, neck and back, may be associated with a condition termed neurocutaneous melanosis ( NCM ). NCM, as well as melanoma risk, appears to be more common in patients with greater numbers of satellite nevi. Patients with NCM have a proliferation of nevus cells in the central nervous system (leptomeningeal melanocytosis), and are predisposed to seizures, MM of the central nervous system (CNS), and neurologic symptoms related to increased intracranial pressure or spinal cord compression. They often seek treatment for symptoms during the first 3 years of life, including lethargy, irritability, headache, recurrent vomiting, seizures, increased head circumference, bulging anterior fontanel, photophobia, papilledema, neck stiffness, and occasionally nerve palsies, particularly of cranial nerves VI and VII. Magnetic resonance imaging (MRI) of NCM reveals focal areas of high signal on T1-weighted images in one or multiple areas of the brain, including the temporal lobes, cerebellum, pons, and medulla. T2 shortening may also occur. In one study, 45% of neurologically asymptomatic children with giant congenital nevi had these radiologic findings. However, a questionnaire-based study of 186 patients with large congenital nevi who were imaged revealed that only 4.8% of those with positive MRI findings for NCM were asymptomatic. Hence the exact prevalence of asymptomatic NCM remains unclear. Other findings in children with NCM may include dorsal spinal arachnoid cysts and amygdalar neuromelanosis (that may be associated with intractable seizures). Overall, the prognosis fo r symptomatic NCM is poor, with more than 90% of patients dying of the disease and around 70% of those dying before 10 years of age.
The management of CMN must be individualized for each patient. There are many factors that must be considered in the decision-making process regarding surgical excision of such lesions. These include location of the nevus, size, cosmetic issues (and potential psychosocial ramifications), and the risks of anesthesia, MM, and neurocutaneous melanosis. There appears to be less consensus regarding the role of surgical excision of small and medium-sized CMN than that of large lesions. Small congenital nevi with uniform pigmentation, smooth texture, and lack of nodules can be clinically monitored, whereas lesions with atypical or deep pigmentation or uneven textures may warrant surgical excision given the heightened difficulty of melanoma detection in this setting. Other risk factors, such as a strong family history of MM or the presence of numerous atypical-appearing nevi, may influence the decision regarding surgical removal of small and medium-sized CMN. Lesions located in areas that may be difficult to follow clinically, such as the scalp or groin, may be better served by excision, although in the case of benign-appearing lesions, this decision may be delayed until the child is older and the procedure can be performed under local anesthesia.
Large CMN are often treated with full-thickness surgical excision. The primary reason for this recommendation is the potentially decreased risk of malignant transformation, although controlled studies supporting this hypothesis are lacking. Unfortunately, excision of large lesions does not usually result in complete removal of all nevus cells, and melanoma may still develop from this residual tissue. In addition, melanoma may develop with increased incidence at extracutaneous sites in these patients, and the significance of neurocutaneous melanosis (if present) must also be factored into this decision. Nonetheless, most still advocate for surgical excision in an effort to reduce the risk of malignancy. Removal of these lesions has been refined with the use of tissue expanders and skin grafting, but multiple procedures are usually required. Partial thickness removal techniques (i.e., dermabrasion, dermatome excision, curettage, chemical peels and laser therapy) have been advocated by some, but the impact of these procedures on malignant transformation or clinical surveillance of the lesion must be further defined. They may, however, be reasonable considerations when more aggressive procedures are impractical. There is no consensus on the timing of surgical procedures for large and giant CMN. Although many experts advocate for early referral and initiation of excisional therapy, other considerations may come into play, including anesthesia concerns surrounding neurocognitive effects on the developing brain. Ongoing prospective studies in this arena will hopefully clarify the optimal use and timing of sedation and anesthesia in young children.
Close periodic clinical surveillance is important for all patients with CMN. Parent education in the importance of sun protection, sunscreen use, and danger signs of atypical nevi or melanoma (see below) is vital. Risks of malignant transformation should be discussed, and parents and patients should be allowed to reach an informed decision regarding therapy. Educational materials regarding moles, melanoma, and sun protection are useful and may be obtained from the American Academy of Dermatology ( www.aad.org ). A multidisciplinary approach must be employed for families of children with large congenital nevi. This includes the primary care physician, dermatologist, plastic surgeon, and diagnostic radiologist. Emotional support should be provided, and the family should be given information on support groups. One such organization is Nevus Outreach ( www.nevus.org ), which was founded by parents of children with large CMN and offers an annual family conference, newsletters, and a social support network.
Atypical Nevi/Familial Atypical Multiple Mole–Melanoma Syndrome
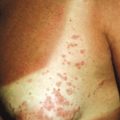
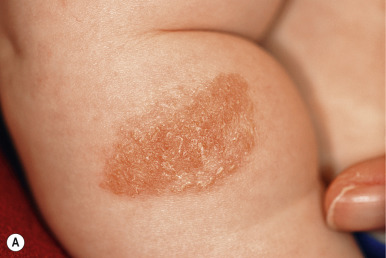
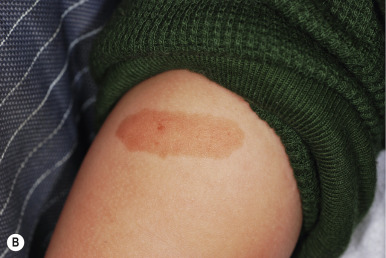
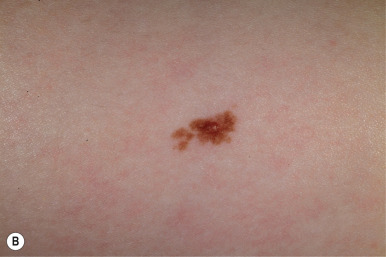
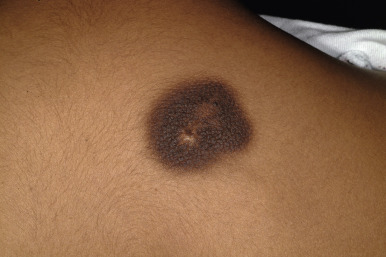
Atypical nevi, also known as dysplastic nevi, are defined based on their clinical and/or histologic appearance. These acquired nevi, which often do not present until at or after puberty, are regarded as markers and potential precursors for MM. The incidence of histologically confirmed atypical nevi in patients under 18 years of age is very low, although in children who come from melanoma-prone families, their incidence seems much higher. Clinically these lesions share some or all of the features of MM ( Box 9-1 ). These include larger size and irregularities of color, texture, or borders ( Fig. 9-16 ). Atypical nevi are often larger than common acquired nevi, usually measuring 6 to 15 mm in diameter, and display marked lesion-to-lesion variability, often with a cobblestone appearance or a dark, central papular component surrounded by a lighter tan flatter periphery (the “fried egg” appearance, Fig. 9-17 ). Other patterns of clinically atypical nevi include the eclipse nevus (tan to hypopigmented center with a darker brown peripheral rim and irregular outer border) and the cockade nevus (target-like morphology with a central pigmented region, an intervening hypopigmented area, and an outer rim of pigmentation; Fig. 9-18 ). These patterns may occur more with greater incidence on the scalp of children (see below).
Familial atypical multiple mole–melanoma (FAMMM) syndrome
Xeroderma pigmentosum
Congenital melanocytic nevi
Atypical (dysplastic) nevi
Personal history of melanoma
Family history of melanoma
High numbers of melanocytic nevi
Fair complexion
Excessive sun exposures
History of blistering sunburns
History of immunosuppression
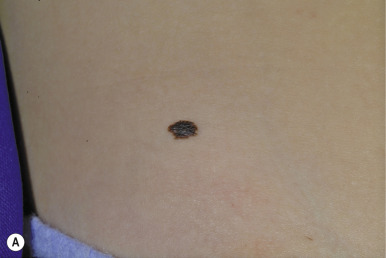
Atypical nevi may occur anywhere, especially on sun-exposed sites, but also commonly occur on covered areas such as the back and in unusual locations such as the buttocks, breasts, and scalp. In fact, a fairly high proportion of nevi removed from the scalp may demonstrate histologic features of atypical nevi. Individuals with atypical nevi have been demonstrated to have an increased risk of melanoma despite the presence or absence of a family history of melanoma. A distinct clinical phenotype characterized by numerous (>100) small, darkly pigmented atypical nevi has been described and termed the cheetah phenotype. Longitudinal follow-up study of such patients is very challenging given the similar clinical appearance of several histologic patterns ranging from benign to cytologically atypical.
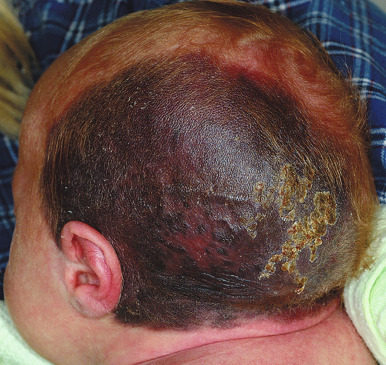
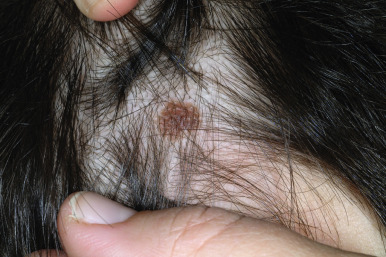
Scalp nevi in children deserve special mention, as they may be a source of anxiety for parents and clinicians alike. These lesions, which are more common in boys, tend to be somewhat larger in size and may often reveal irregular borders and some color variations ( Fig. 9-19 ). The observed patterns may include the fried-egg, cockade, and eclipse phenotypes discussed previously, as well as clinically benign nevi patterns. Although scalp melanoma does occur in children, it seems that the majority of these lesions are benign; however, they may reveal mild to moderate atypia microscopically. Dermatoscopic (magnified light) examination may be helpful in demonstrating benign patterns (i.e., globular or reticular–globular pigmentation and perifollicular hypopigmentation). It should be remembered that although the majority of scalp nevi in children are benign, their presence may suggest an increased risk for the development of numerous melanocytic nevi, which itself is a risk factor for MM.
The familial atypical multiple mole–melanoma (FAMMM) syndrome is a disorder of autosomal dominant inheritance in which atypical nevi develop during the late second to third decade of life. It was originally described in 1978 by Clark and colleagues when they noted individuals with increased numbers of melanocytic nevi that displayed atypical clinical and histologic features. Since the original description, FAMMM syndrome has been better characterized as a hereditary cancer predisposition syndrome, entailing not only an increased risk of MM but also other cancers in some kindreds, especially pancreatic cancer. FAMMM syndrome is characterized by the familial occurrence of melanoma in combination with multiple dysplastic nevi (>50) in first-degree relatives, although around 10% of the melanoma patients may have very few to no dysplastic nevi. It has been estimated that individuals with a family history of FAMMM syndrome and atypical nevi have nearly a 100% lifetime risk of acquiring MM. In addition to melanoma of the skin, these individuals may develop intraocular melanoma. Recent observations of families displaying the association of pancreatic cancer and melanoma have led to the alternative terminology of FAMMM–pancreatic cancer ( FAMMM–PC ) syndrome . The phenotype of these families is variable, however, with several reported members lacking the dysplastic nevus phenotype. Some have therefore suggested the possibility of two distinct hereditary cancer syndromes, FAMMM–PC and the pancreatic cancer/melanoma syndrome (PCMS).
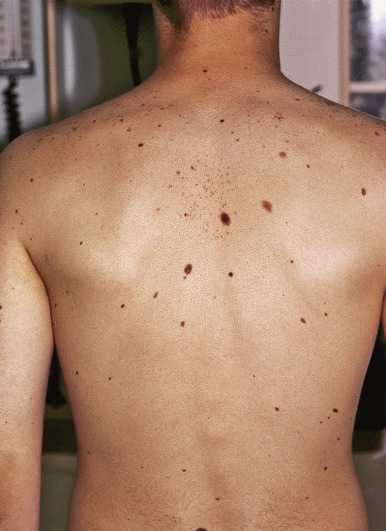
The classic features of FAMMM syndrome are high numbers of melanocytic nevi, often more than 50, some of which are atypical and often have large variability in size ( Fig. 9-20 ), that occur in the setting of a family history of MM in one or more first- or second-degree relatives. Identification of at-risk children is vital, because melanoma may occur before 20 years of age in up to 10% of individuals. Although most of the atypical nevi develop during or after adolescence, prepubertal children may occasionally show the atypical nevus phenotype, particularly in scalp nevi. In fact, the development of scalp nevi during childhood, as well as large nevus counts early in life, may represent early indicators of increased risk for the atypical mole phenotype later in life.
There are several known melanoma susceptibility loci, most notably the p16 CDKN2A gene on chromosome 9, which is a tumor-suppressor gene responsible for melanoma susceptibility in some kindreds with FAMMM syndrome. Deletion of p16 may play a role in the development of atypical nevi as an early event, as well as in the development of MM (see below).
Management of the patient with atypical nevi should include a thorough family history, total body inspection, and a regular clinical follow-up examination every 6 to 12 months, depending on the individual patient. Patients (and parents) should be educated regarding sun protection, regular skin self-examination, and atypical features of nevi, and surgical excision should be performed for lesions with concerning features. A helpful guideline in monitoring children with many nevi is the “ugly duckling” principle, which dictates that a lesion that stands out from the rest (i.e., markedly darker pigmentation, more irregularity of texture or borders) should be more closely scrutinized and considered for removal. Epiluminescence microscopy (dermatoscopy) is a useful noninvasive instrument that provides magnification of nevi and may be useful in differentiating benign from atypical lesions. It is most often used by dermatologists, and its use requires training and experience in order to provide acceptable sensitivity and reliability. Serial photography of nevi may also be a useful adjunct in the longitudinal follow-up study of patients with multiple or atypical nevi.
Malignant Melanoma
The incidence of MM, the most deadly form of skin cancer, continues to rise, and although childhood MM continues to be rare, the incidence in this population may also be rising. The lifetime incidence of melanoma is estimated at around 1 in 58 individuals, which is an alarming increase from the incidence of 1 in 1500 estimated in 1935. The challenges of diagnosing MM in children are multiple and include lack of recognition, hesitancy to perform skin biopsies in children, and poor reliability in making the histopathologic diagnosis in this population. Melanoma in individuals under 20 years of age accounts for only 1% to 3% of all MM, and prepubertal cases are even rarer, accounting for 0.3% to 0.4% of all cases. Despite this rarity, the course of melanoma, when it does occur in a child, bears the same prognosis as it does in adults. Importantly, the incidence of pediatric melanoma is increasing at an estimated 1% to 4% per year. Interestingly, many lesions previously thought to be melanomas of childhood are now recognized to be benign Spitz nevi (see below). Congenital MM represents a small subset of all pediatric MM and is extremely rare, with only 23 cases reported in the English literature between 1925 and 2002. It most commonly arises in a congenital melanocytic nevus and less commonly is de novo or related to transplacental transmission of MM from the mother.
MM most commonly affects individuals with fair skin, blue eyes, and red or blond hair, particularly those of Celtic origin whose pigment cells have a limited capacity to synthesize melanin. Approximately one-half to 65% of MM arises in a preexisting nevus. Although melanomas may occur in African-American individuals, the incidence is extremely low when compared with that of Caucasians. In African-Americans, tumors usually arise in areas that are lightly pigmented, especially the mucous membranes, nailbeds, or the sides of the palms and soles. Exposure to high levels of sunlight in childhood seems to be a strong determinant of risk for MM, although adult sun exposure also plays a role. Some studies have not supported this “critical risk period” hypothesis of ultraviolet radiation exposure, suggesting that sun protection education should be directed at the entire population with equal effort rather than concentrating solely on younger age groups. However, maintaining a stringent focus on childhood sun protection still seems reasonable, because a large proportion of overall lifetime sun exposure is likely to occur during these years.
The genetics of MM continue to be elucidated, and it is now established that molecular defects in both tumor-suppressor genes and oncogenes may be pathogenetically linked to melanoma. Mutations in the CDKN2A (p16) gene predispose a patient to the FAMMM syndrome and have also been noted in families in which only one or two individuals are affected by MM. A subset of melanoma-prone families with CDKN2A mutations also manifests an increased risk of pancreatic cancers (as discussed earlier). Activating mutations in one of two Ras/mitogen-activated protein kinase pathway genes, BRAF or NRAS , are present in up to 80% to 90% of melanomas. Some of the other genes potentially mutated in melanoma include PTEN , CDK4, CCND1, KIT, p53, EphA2 , ERBB4, GRIN2A, GRM3, MEK1, MEK2, MITF , β-catenin , and various apoptosis genes.
Melanoma is classically categorized according to its clinical and histologic features. The most common forms are superficial spreading, nodular, lentigo maligna (usually confined to adults), and acral lentiginous melanoma. However, pediatric cases of MM often cannot be neatly classified into one of these categories. Pediatric melanoma seems to follow the same distribution patterns as adult melanoma, with head and trunk lesions predominating in boys, and arm and leg lesions predominating in girls. Risk factors for pediatric MM are listed in Box 9-1 .
The classic signs of MM are summarized in Table 9-1 . Melanoma typically presents with a rapidly enlarging papule or nodule, most often brown to brown-black in color ( Fig. 9-21 ), although blue, red, or white discoloration may also be noted. Importantly, a significant proportion of pediatric melanomas may present in an amelanotic (i.e., pink, white, red, or a combination of these colors) fashion. A halo of hypopigmentation or depigmentation may be present around a primary lesion of MM, although most often these halos occur in the setting of benign nevi (see Halo Nevus section). Melanomas often reveal asymmetry and irregularity of the borders, especially scalloping or notching, and tend to be larger than benign nevi, often (but not always) larger than 10 mm in diameter. Bleeding, itching, ulceration, crusting, and pain may be present. In a recent single-center retrospective review of 70 children (<20 years of age) with melanoma or other atypical melanocytic tumors, several clinical features were highlighted that may contradict the traditional “ABCD” portion of the ABCDE criteria (as shown in Table 9-1 ). These include the amelanotic (flesh-colored, pink, or red) nature of the lesions, bleeding, uniformity of color, small diameter (often <6 mm) and de novo development (as opposed to arising in an existing nevus). These characteristics should be kept in mind when evaluating children with atypical “bumps” and may facilitate earlier recognition of (and hence therapy for) pediatric MM.
| Clinical Feature * | Comment | |
|---|---|---|
| A | Asymmetry | The two halves of the lesion are not alike |
| B | Border irregularity | Borders notched, scalloped, irregular |
| C | Color changes | Especially blue, red, black, white |
| D | Diameter >6 mm | Size of a pencil eraser; not applicable to congenital nevi, which are often >6 mm early in their evolution |
| E | Enlargement | Evolutionary change in the lesion |
* See text for important discussion of features that may be more specific to pediatric melanoma.
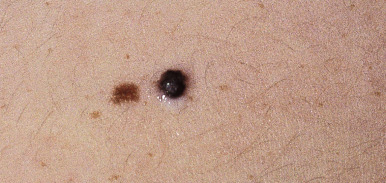
Lymph nodes may be palpable and if present, are an ominous prognostic sign, suggesting metastatic spread. When MM goes undetected and undiagnosed, the lesion proliferates locally and may spread by satellite lesions or extend through the lymphatics or bloodstream, from which it may eventually invade any organ of the body. Once metastatic disease occurs, the prognosis of MM significantly declines.
Early detection of melanoma is germane to long-term survival. The survival rate for children and adults seems to be similar, and the primary determinants of prognosis have traditionally been tumor thickness and depth of invasion. The presence or absence of ulceration has been added to the American Joint Committee on Cancer (AJCC) staging system for MM as another important prognostic criterion (the presence of ulceration correlating with poorer prognosis). The pediatrician (or other primary care provider of children) is in good position to observe pigmented skin lesions over time in their patients. Early referral should be considered for any melanocytic lesion that displays substantial growth changes, especially if asymmetry, ulceration, or other atypical features are present. Melanoma is diagnosed by histopathologic examination of skin tissue from biopsy. When MM is suspected, full-thickness excision of the lesion should be performed, as depth of the lesion must be fully visualized in order to assess this prognostic indicator. Shave biopsies or excisions should be avoided in the removal of concerning pigmented lesions.
Surgical excision is the initial step in melanoma management, along with adequately staging the disease. Once the diagnosis of MM is confirmed, the site is reexcised with appropriate margins (0.5 to 3 cm), depending on tumor depth in the initial specimen. Clinically suspected lymph nodes are surgically removed and histologically evaluated. Developments in tracer techniques have enabled sampling of the first lymph node draining the affected skin site (the “sentinel” lymph node), which assists in determining whether to proceed with regional lymph node dissection for further staging and therapy. This technique, which is usually performed concurrently with reexcision of the primary lesion, allows for accurate staging of the regional lymph nodes with minimal morbidity. In a series of 126 patients younger than 21 years of age who were diagnosed with MM, sentinel lymph node metastases were present in 29%, yet the survival was equal to or better than that reported for adults, suggesting that the biology of pediatric melanoma may be different than that in adult melanoma. It appears that lymph node metastases and thicker tumors may be more common in younger children (<10 years of age) with MM. In addition to histopathologic analysis and lymph node examination, staging for pediatric MM may also include computed tomography (CT), positron emission tomography (PET) and PET-CT scanning in patients with thicker lesions to evaluate for distant metastases.
Treatment of more advanced stages of MM includes chemotherapy, radiotherapy, and immunotherapy, although the results have been somewhat discouraging. Adjuvant interferon α-2b therapy has received much attention in the treatment of high-risk melanoma in adults, with reports of improved disease-free survival and overall survival rates with use of the Kirkwood regimen (high-dose therapy for 4 weeks followed by a lowered dose for 48 weeks). Recommendations for use of this agent in children have primarily been extrapolated from the adult data, and although its use is potentially effective, it may be associated with some toxicities at higher doses, which may be limiting when treating the pediatric melanoma patient. Immunotherapeutic approaches to MM include melanoma vaccines, cytokine therapy, and passive immunotherapy with monoclonal antibodies, and these and other approaches continue to be the topics of active investigation. Traditional chemotherapy regimens have shown minimal activity against melanoma. Biologic agents such as high-dose interleukin (IL)-2 have resulted in durable responses in some adult patients, but experience in children is limited. More recently, treatments that target growth-factor pathways or molecular targets have received increasing attention, including BRAF inhibitors such as dabrafenib and vemurafenib, and MEK inhibitors such as trametinib. The combination of BRAF and MEK inhibition appears to provide greater benefit than BRAF inhibitor monotherapy in adults. These treatments have not yet been well studied in pediatric melanoma.
The prognosis for pediatric melanoma is strongly correlated with initial stage. In a review of data from the National Cancer Database of 3158 patients aged 1 to 19 years with melanoma, average 5-year survival rates were 98.7% ( in situ disease), 93.6% (localized invasive disease), 68% (regionally metastatic disease), and 11.8% (distant disease). Some studies have found decreased survival in prepubescent patients with MM, whereas others have found no difference or a better prognosis in this group. Hence there still exists no consensus on the overall prognosis for pediatric MM.
Spitz Nevus
Spitz nevus is a distinct subtype of melanocytic nevus that occurs primarily in children. The importance of the Spitz nevus, which was formerly known as benign juvenile melanoma, lies in its histologic differentiation from MM. This lesion was originally recognized in 1948 when Dr. Sophie Spitz realized that a subset of juvenile melanomas did not behave in the same fashion as adult melanomas. Spitz nevi have subsequently been identified as a distinct nevus variant, occurring most commonly on the face of children and adolescents.
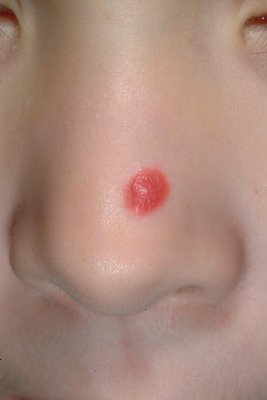
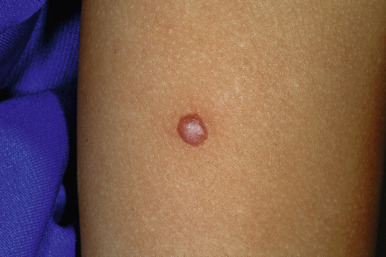
Spitz nevi present usually as a smooth-surfaced, hairless, dome-shaped papule or nodule with a distinctive red-brown color ( Fig. 9-22 ). They are most often solitary, although multiple clustered (agminated, Fig. 9-23 ) or disseminated lesions have been described. They may vary in size from a few millimeters to several centimeters, although most range from 0.6 to 1 cm in diameter. The lesion may be so red that the differential diagnosis for some includes pyogenic granuloma or early juvenile xanthogranuloma. The presence of brown pigmentation ( Fig. 9-24 ), either with regular clinical examination or when examined through a glass slide compressing the surface (diascopy), may be useful in confirming the melanocytic nature of the lesion. Surface telangiectasia may also be a prominent feature. In some lesions, particularly those on the extremities, the red color is replaced by a mottled brown to tan or black appearance, often with verrucous surface changes and irregular borders. Clinically it is this type of lesion that is most easily confused with MM ( Fig. 9-25 ).
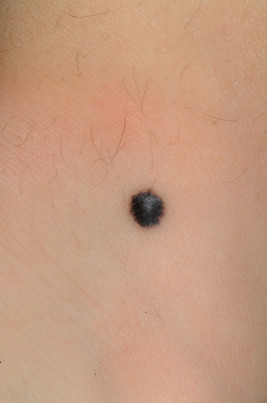
Although most Spitz nevi behave in a benign fashion, local recurrence after excision may occur in as many as 5%. It has been suggested by some that occasional Spitz nevi are malignant in origin and have the potential for more aggressive biologic behavior. Whether these represent a subtype of Spitz nevus or a “spitzoid” MM is unclear. It is obvious that some Spitz nevus-like lesions may pose substantial diagnostic difficulties especially when atypical features are present, even among dermatopathology experts. These lesions have been variably called spitzoid tumor of uncertain malignant potential or atypical spitzoid tumor. A grading system has been proposed for Spitz nevi with atypical features, and application of such a system may be useful in guiding management for patients with atypical Spitz tumors. BRAF mutations have occasionally been observed in a subset of Spitz nevi, suggesting that this finding should not be relied upon for distinguishing Spitz nevus from melanoma. The expression of cell cycle and apoptosis regulators in Spitz nevi appears to more closely parallel the findings in benign nevi rather than melanoma. More recently, fluorescent in situ hybridization (FISH) using probes for chromosomal loci has been used in an attempt to predict biologic behavior in atypical spitzoid tumors; gains in 6p25 or 11q13 and deletions in 9q21 have been shown to correlate with more aggressive clinical behavior. Deletions in 9q21 appear to correlate most with lymph node spread and distant metastasis.
The management of Spitz nevi is controversial. Many experts recommend excision of these lesions on the basis of uncertainty in their biologic behavior, occasional reports of aggressive potential, and the increasing observation of the amelanotic nature of many melanomas in children. Others advocate for watchful waiting, reserving excision for lesions that demonstrate atypical features or those of psychosocial significance. When these lesions are excised, however, complete removal is advisable. More importantly, surgical specimens should be examined by a dermatopathologist or pathologist experienced in the diagnosis of melanocytic lesions and familiar with Spitz nevi. If an excised lesion is diagnosed unequivocally as a Spitz nevus without atypical features, no further therapy is necessary. However, because incompletely removed lesions may recur and result in a histopathologic appearance that may be more likely to be misinterpreted as MM, conservative reexcision is recommended for lesions with positive margins noted on the initial biopsy.
Halo Nevus
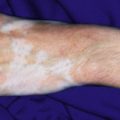
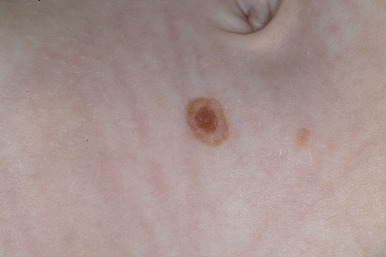
A halo nevus is a unique skin lesion in which a centrally placed, usually pigmented nevus becomes surrounded by a 1- to 5-mm halo of hypopigmentation or depigmentation ( Fig. 9-26 ). These lesions are common in children and young adults. The cause of the spontaneous loss of pigmentation is unknown but appears to be related to an immunologic destruction of melanocytes and nevus cells. Adding support to this hypothesis is the fact that several patients with halo nevi have a tendency toward the development of vitiligo (see Chapter 11 ; Fig. 9-27 ). Histologic examination of halo nevi reveals reduction or absence of melanin and a dense inflammatory infiltrate around the central nevus. Although compound or intradermal nevi are the tumors most commonly associated with the halo phenomenon, it may also occur around blue nevi, Spitz nevi, neurofibromas, melanomas, and metastatic lesions of melanoma. Giant CMN may also reveal the halo phenomenon with pigment regression and at times, self-destruction.
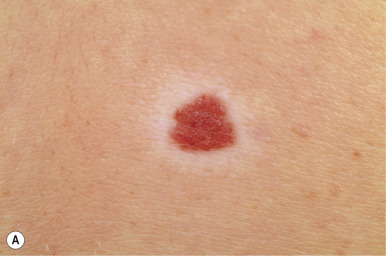
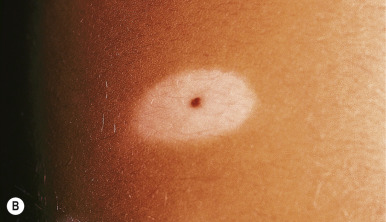
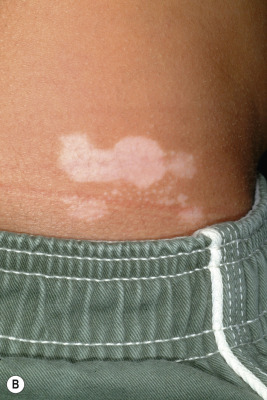
Typical halo nevi are notable for loss of pigmentation in the nevus, with a pink appearance and commonly, eventual disappearance of the original melanocytic lesion. Occasionally, darkening of the central nevus may occur. Halo nevi may appear on almost any cutaneous surface, but the site of predilection for most lesions is the trunk, particularly the back. In most patients, eventual repigmentation of the halo occurs over a period of months to years.
Halo nevi tend to be benign, although the halo phenomenon may occur around lesions revealing varying degrees of histologic atypia. Potential concern has been raised over reports of MM exhibiting the halo phenomenon and the increased incidence of halo nevi in adults with melanoma. In a survey of pediatric dermatologists, no diagnoses of MM in pediatric patients with halo nevi were noted. Clinical features that may suggest an increased probability of an atypical melanocytic lesion within a halo include the ABCDE diagnostic criteria of melanoma (see Table 9-1 ) and asymmetry or irregularity of the surrounding depigmentation. Any patient with a halo nevus, especially if multiple halo lesions are present, should receive a complete skin and mucous membrane examination to assess for melanocytic lesions revealing atypical features. Patients with the halo nevus phenomenon, concomitant vitiligo, and ocular melanoma have been described, but in general ophthalmologic evaluation is not routinely indicated. If the melanocytic lesion in the central portion of a halo reveals concerning or atypical features, complete excision should be performed. If, on the other hand, the central lesion has benign characteristics, excision is unnecessary and the lesion may be observed at intervals until it has resolved.
Nevus Spilus
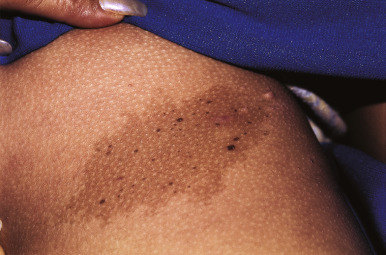
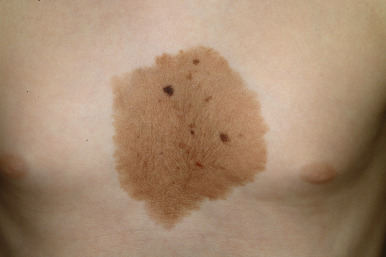
Nevus spilus is a solitary, nonhairy, flat, brown patch of melanization dotted by smaller dark brown to black macules ( Fig. 9-28 ). This relatively common lesion, although usually present at birth, may first become noticed during infancy, childhood, or even later. However, clinical and histologic data suggest that these lesions are most likely a subtype of CMN. The earliest findings are usually similar to a café-au-lait patch, with eventual development of the secondary superimposed darker melanocytic lesions. Nevi spili may vary in size from 1 to 20 cm in diameter and may appear on any area of the face, trunk, or extremities without relation to sun exposure. Subtle hypertrichosis may occasionally be present ( Fig. 9-29 ). Although the darker melanocytic components of these lesions have the potential to develop MM, the incidence of this transformation appears to be low. Histologic evaluation of a nevus spilus usually reveals components of junctional and congenital nevi. Patients with nevus spilus should be monitored longitudinally with serial clinical examinations and if possible, photographic surveillance. Any areas revealing atypical clinical features should be selectively excised and subjected to histologic evaluation, but widespread prophylactic excision seems unwarranted. These lesions (especially when larger) have also been called speckled lentiginous nevi ( SLN ). Although they usually occur in isolation, they may sometimes be associated with other organ abnormalities as part of a syndrome such as phakomatosis pigmentovascularis (see Chapter 12 ), phakomatosis pigmentokeratotica (PPK), or SLN syndrome (see below).
Becker Melanosis
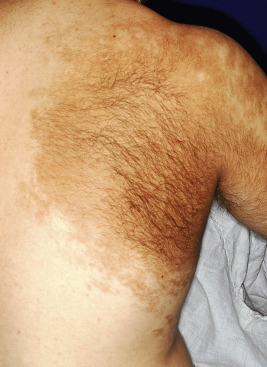
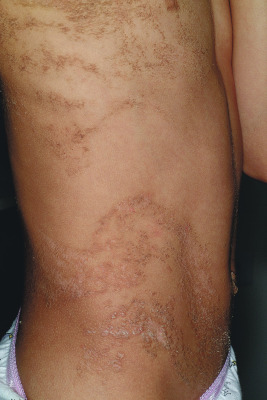
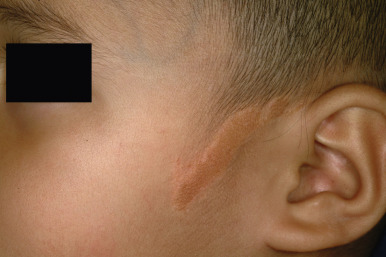
Becker melanosis, also known as Becker nevus, is an acquired, unilateral hyperpigmentation usually involving the upper trunk of adolescent males. Occasionally it may present very early in life (as early as birth) and may be distributed in other locations, including the extremities. This pigmentation, which is caused by increased melanization of the epidermis and not by nevocellular proliferation, may be associated with hypertrichosis ( Fig. 9-30 ) and occasionally proliferation of smooth muscle derived from erector pili muscles. Becker melanosis is discussed in more detail in Chapter 11 .
Tumors of the Epidermis
Tumors of the epidermis range in spectrum from benign lesions to those that are malignant. Benign tumors appear much more often than malignant lesions in children, and the latter, when they do occur, may be overlooked and/or the diagnosis delayed.
Epidermal Nevi
Epidermal nevi (EN) are benign congenital lesions characterized by hyperplasia of epidermal structures. They are usually apparent at birth or become noticeable during early childhood, affect both sexes equally, and are known by several descriptive names, including nevus verrucosus, nevus unius lateris, and ichthyosis hystrix. In addition, EN can be divided into nonorganoid (keratinocytic) nevi and organoid EN, such as nevus sebaceous or follicular nevi. Although the exact etiology of EN is unknown, activating fibroblast growth factor receptor 3 (FGFR3) mutations have been demonstrated in some, as have mutations in the p110 α-subunit of PI3K ( PIK3CA ), HRAS, KRAS, and NRAS .
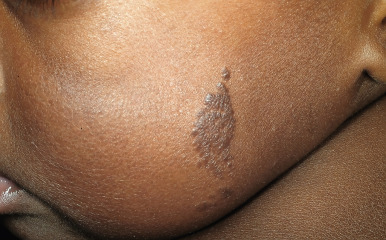
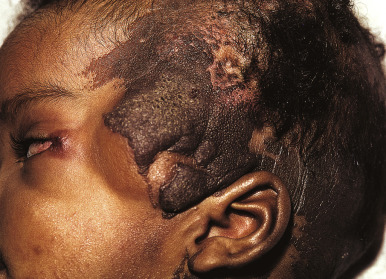
Keratinocytic EN (often referred to simply as EN ) may be slightly or darkly pigmented and unilateral or bilateral in distribution. They often favor the extremities, although they may occur anywhere on the cutaneous surface. EN are usually distributed in a mosaic pattern of alternating stripes of involved and uninvolved skin. This pattern is termed Blaschko lines and occurs as a result of migration of skin cells during embryogenesis. Disorders that occur along the Blaschko lines usually reveal a linear pattern on the extremities and a wavy or arcuate pattern on the trunk. Although a single EN is most common, multiple lesions may be present, sometimes in association with the epidermal nevus syndrome (ENS; see below). The localized form is often present at birth and presents as a tan to brown, velvety or verrucous (warty) papule or plaque. There may be a single lesion ( Fig. 9-31 ) or multiple lesions, and a linear configuration is common ( Fig. 9-32 ). One subtype of epidermal nevus has been termed the acanthosis nigricans form of EN and is characterized by a clinical ( Fig. 9-33 ) and histologic resemblance to acanthosis nigricans.
The term nevus unius lateris has been used traditionally to describe extensive unilateral lesions. Nevus unius lateris may present as a single or spiral linear, verrucous lesion or at times as an elaborate, continuous, or interrupted pattern affecting multiple sites ( Fig. 9-34 ) and occasionally involves more than half of the body. Systematized epidermal nevus has been used to describe extensive lesions that are bilateral and in which truncal involvement predominates.
EN may reveal a variety of histologic features. Importantly, those that reveal epidermolytic hyperkeratosis, a distinct pattern of clumping of keratin filaments in the suprabasal cells of the epidermis, imply a mosaic disorder of keratin genes. Patients with this condition, especially when skin involvement is extensive, may transmit these mutations to offspring, resulting in a more widespread ichthyosiform condition termed epidermolytic ichthyosis and epidermolytic hyperkeratosis (also known as bullous congenital ichthyosiform erythroderma; see Chapter 5 ). These EN may be clinically indistinguishable from other EN ( Fig. 9-35 ).
EN are challenging to treat, given the observation that most superficial destructive therapies are followed by recurrence of the lesions. These superficial therapies have included cryotherapy with liquid nitrogen, dermabrasion, electrodesiccation, and laser ablation. In a series of 71 treated lesions, cryotherapy of small EN resulted in an excellent response in 90%. Carbon dioxide (CO 2 ) laser therapy may offer excellent results, but response to therapy is unpredictable. Staged CO 2 laser ablation has been used successfully, both with and without preceding surgical debulking. Topical therapies used with variable success include retinoids, 5-fluorouracil, steroids, and podophyllin, among others. Photodynamic therapy with methyl-aminolevulinate was reportedly successful in a young girl, although hypertrophic scarring occurred. Full-thickness surgical excision or deeper destructive procedures (such as deep dermabrasion) appear effective at removing these hamartomas but are generally limited to smaller, more localized lesions. Since these lesions may continue to extend during childhood, surgical intervention should be delayed until the full extent of the process is determined.
Epidermal Nevus Syndrome
ENS is a sporadic association of EN with abnormalities in other organ systems. Some believe that this syndrome is actually a group of several syndromes, each with distinguishing cutaneous and extracutaneous features. Happle suggests that the five well-defined ENSs are Schimmelpenning syndrome, nevus comedonicus syndrome, pigmented hairy ENS, Proteus syndrome (see Chapter 12 ), and congenital hemidysplasia with ichthyosiform nevus and limb defects (CHILD) syndrome (see Chapter 5 ). Keratinocytic ENS also merits inclusion on this list. Regardless, patients who fall into the spectrum of having an ENS generally have organoid or nonorganoid EN in conjunction with defects in the CNS, eyes, musculoskeletal system, connective tissue and occasionally other organ systems. The manifestations of ENS are believed to represent genomic mosaicism with the effects of the genetic defect(s) and timing of the mutation during development determining the spectrum of clinical involvement.
Phakomatosis pigmentokeratotica ( PPK ) has been used to describe the association of speckled lentiginous nevus with an epidermal nevus that has sebaceous differentiation and is accompanied by skeletal and neurologic abnormalities. It has been suggested that patients with PPK may have systemic features suggestive of either Schimmelpenning syndrome (extensive sebaceus nevi, mental retardation, seizures, coloboma, lipodermoids of the conjunctiva, skeletal defects, and vascular abnormalities) or what has been termed SLN syndrome (presenting with SLN, hyperhidrosis, sensory and motor neuropathy, nerve palsy, and spinal muscular atrophy).
Examples of extracutaneous abnormalities seen in patients with ENS include seizures, mental retardation, hemiparesis, hypotonia, cranial nerve palsies, developmental delay, deafness, kyphosis, scoliosis, limb hypertrophy, hemihypertrophy, facial bone deformity, macrocephaly, ocular lipodermoids, coloboma, corneal changes, cortical blindness, cataracts, and retinal changes. CNS imaging may reveal hemimegalencephaly, agenesis of the corpus callosum, cerebral heterotopia, cortical agyria or microgyria, or Dandy–Walker malformation; brainstem and cerebellar malformations and neonatal medulloblastoma have also been reported. In addition, hypophosphatemic vitamin D-resistant rickets has been observed in patients with ENS, and in some reports the hypophosphatemia improved after surgical revision of the EN. It has been suggested that some EN may produce a phosphaturic substance that contributes to this association. Hyponatremia has also been observed as an early manifestation in a patient with ENS. Cardiac and genitourinary defects may also be associated.
The features of the cutaneous lesions in ENS range from large unilateral nevus unius lateris-like lesions to diffusely distributed, whorled lesions ( Figs. 9-36 and 9-37 ) involving variable degrees of the skin surface or linear, orange-yellow plaques as seen in nevus sebaceous of Jadassohn (see Nevus Sebaceous section). Plaques of dilated follicular pits filled with keratin may be seen in the nevus comedonicus type of ENS, and extensive Becker nevus is seen in the pigmented hairy type of ENS. In the CHILD syndrome, a unilateral inflammatory epidermal nevus with a sharp midline demarcation and an affinity for body folds is seen in conjunction with the characteristic ipsilateral hypoplasia of limbs and other organ defects. Patients with Proteus syndrome have verrucous EN in association with partial gigantism, macrocephaly, and vascular malformations.
Patients with large or extensive EN require careful medical, family, and developmental histories and thorough physical evaluation, with particular emphasis on the musculoskeletal, neurologic, ocular, and cardiovascular examinations. Management of ENS should be multidisciplinary including a dermatologist, pediatrician, neurologist, ophthalmologist, and plastic surgeon, with utilization of other subspecialists as necessary. EN can be treated as noted earlier, although treatment is even more challenging given their extensiveness in this setting. Malignant transformation of EN is rare but may occur, both in syndromic and nonsyndromic lesions, and includes basal cell carcinoma (BCC) or squamous cell carcinoma (SCC), depending on the type of epidermal nevus.
Inflammatory Linear Verrucous Epidermal Nevi
Inflammatory linear verrucous epidermal nevi (ILVEN) appear to be a unique variant of epidermal nevus that presents as a chronic pruritic process with erythematous, scaly, and verrucous papules that coalesce into linear plaques ( Fig. 9-38 ). These lesions are often present at birth or may appear during early childhood and most often occur on an extremity. They are notable for their chronic and intermittent course and resistance to therapy. Occasionally lesions may spontaneously improve or resolve only to eventually reappear. The differential diagnosis of ILVEN includes linear psoriasis, lichen striatus, linear lichen planus, and verrucous epidermal nevus. ILVEN can usually be confirmed by the clinical course, morphologic appearance of lesions, intense pruritus, and resistance to therapy.
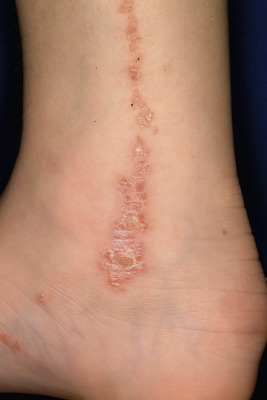
Treatment of ILVEN is difficult, as discussed previously for EN. Topical or intralesional corticosteroids may reduce inflammation and pruritus and produce a temporary remission, but the lesions generally recur. Topical retinoids, 5-fluorouracil, calcineurin inhibitors (i.e., tacrolimus or pimecrolimus), CO 2 laser therapy, and vitamin-D derivatives (i.e., calcitriol) have been used with varying results. Photodynamic therapy with methyl-aminolevulinate was reportedly successful in an adult with ILVEN. Patients with extensive and symptomatic ILVEN have been treated surgically, with tissue expansion, serial full-thickness excisions, and split-thickness skin grafting with excellent outcomes.
Basal Cell Carcinoma
Basal cell carcinoma (BCC) is a slow-growing, usually nonmetastasizing but invasive malignant skin tumor with varying clinical patterns that may be triggered by ultraviolet radiation exposure. This disorder arises from the basal cells of the epidermis or its appendages and is most commonly seen in persons of middle age. BCC is the most common form of skin cancer, and although rarely seen in children, it can occur in childhood and must be considered even in the very young. However, misdiagnosis must also be considered in a child, as several benign hamartomas of follicular differentiation (trichoepithelioma, trichoblastoma, trichofolliculoma) may histologically appear similar to BCC to the inexperienced pathologist.
When the diagnosis of BCC is confirmed in a child, one must consider an associated predisposing condition such as basal cell nevus syndrome (BCNS, or Gorlin syndrome, see below), xeroderma pigmentosum (see Chapter 19 ), Bazex syndrome, and Rombo syndromes (see Chapter 7 ), albinism (see Chapter 11 ), or an underlying nevus sebaceous of Jadassohn (see Nevus Sebaceous section). Other genetic skin disorders that may entail an increased risk for BCC include Bloom syndrome, Werner syndrome, Rothmund–Thomson syndrome, Muir–Torre syndrome, Brooke–Spiegler syndrome (BSS), Cowden syndrome (CS), and some immunodeficiency disorders. Children treated with irradiation for malignancy or with solid organ transplantation may develop BCC years to decades after the treatment. In a review of childhood cancer survivors who were subsequently diagnosed with BCC, the majority of tumors developed between 20 and 39 years of age, and the risk was noted to be increased when patients received radiation doses to the skin of more than 1 Gy. BCC lesions have occasionally been reported as sporadic cases in children without any underlying predisposition and appear to be most often located on the head, back, and chest. The diagnosis of BCC in childhood is often delayed because of a low index of suspicion.
The majority of BCCs have a predilection for the upper central part of the face. Although they may arise without apparent cause, prolonged exposure to the sun is a predisposing factor, particularly in individuals with a fair skin phenotype. BCC may occur in several clinical forms. Noduloulcerative BCC, the most common type, begins as a small, elevated, translucent papule or nodule with telangiectatic vessels on its surface. It may enlarge, develop central necrosis, and result in an ulceration surrounded by a pearly rolled border. Although this form usually occurs as a single lesion, patients who develop this form of basal cell tumor often are likely to develop other such lesions. Superficial BCC presents as an erythematous, scaly, minimally elevated papule or plaque that may have superficial crusting. Often multiple, these lesions tend to occur on the trunk or extremities, expand slowly, and are easily mistaken for lesions of psoriasis, dermatitis, or tinea. Pigmented BCCs are similar to noduloulcerative lesions but also contain irregular brown pigmentation that may simulate the appearance of a nevus or MM. Sclerosing or morpheaform BCC presents as a firm, yellow-white waxy papule or plaque with an ill-defined border and absence of the translucent rolled edge. Tumors of this type have been known to arise in early childhood and may grow for years before attracting medical attention.
In the pediatric patient who is diagnosed with a BCC, a thorough history and physical examination should be performed with attention to the regional lymph node examination. An evaluation for an associated predisposing condition should be performed when indicated. No single method of therapy is applicable to all BCC lesions. The goal, as with any skin tumor, is for permanent cure with the best functional and cosmetic result. Curettage and electrodesiccation is a simple office therapy most commonly used by dermatologists for low-risk, small BCC in areas without a dense hair pattern. Excision (with or without Mohs micrographic surgery [MMS]) is the treatment of choice for childhood-onset BCC. Radiation therapy can be an effective treatment but is not desirable in children (and contraindicated in the setting of BCNS, discussed later in this chapter, because it may increase the risk for invasive BCC). Other treatment modalities include cryotherapy, CO 2 laser therapy, photodynamic therapy, systemic retinoids, topical chemotherapy (i.e., topical 5-fluorouracil), and biologic-response modifiers. The latter include imiquimod, a topical immune-response modifier demonstrated to be effective against superficial BCCs and small nodular BCCs. This agent promotes innate immune responses and exhibits antitumor as well as antiviral effects, having been initially approved for the treatment of anogenital condylomata. Experience with these therapeutic methods in childhood BCC is primarily anecdotal. Sun protection and skin self-examination education is vital.
Basal Cell Nevus Syndrome
BCNS (also known as Gorlin syndrome or nevoid BCC syndrome ) is an autosomal dominant disorder with complete penetrance and variable expressivity characterized by childhood onset of multiple BCCs and associated with other abnormalities, including odontogenic jaw cysts, bifid ribs, and intracranial calcification. The most obvious cutaneous feature in patients with BCNS is the appearance of multiple BCCs early in life. These basal cell epitheliomas are indistinguishable on histopathologic examination from ordinary BCCs. The diagnostic criteria for BCNS are shown in Box 9-2 . In a consensus statement from an international colloquium on BCNS, a modification of the diagnostic criteria was suggested such that medulloblastoma, typically desmoplastic, be included as a major (rather than a minor) criterion.
* Diagnosed when an individual has two major criteria and one minor criterion, OR one major criterion and three minor criteria.
, †† See text for further discussion.
Major criteria
Multiple basal cell carcinomas (>5 in a lifetime or a BCC before 30 years)
Lamellar calcification of the falx (or clear evidence of calcification in an individual younger than 20 years)
Jaw keratocyst (odontogenic keratocyst, confirmed histologically)
Palmar or plantar pits (>2; may be most easily seen after soaking of the hands and feet in warm water)
First-degree relative with BCNS
Minor criteria
Macrocephaly
Childhood medulloblastoma
Cleft lip/palate
Rib/vertebral anomalies: bifid, splayed, missing or extra ribs; bifid, wedged, or fused vertebrae
Preaxial or postaxial polydactyly
Cardiac or ovarian fibroma
Lymphomesenteric or pleural cysts
Ocular anomalies (cataract, coloboma, microphthalmia)
BCC, Basal cell carcinoma; BCNS, basal cell nevus syndrome.
The skin lesions of BCNS may appear as early as the first year of life but have a mean age of onset of around 20 to 23 years. They involve, in decreasing order of incidence, the face, neck, back, trunk, and upper extremities. The distribution of BCCs may differ by gender, with male patients having more in the facial M-zone (forehead, temples, periorbital areas, and nose), upper back, neck, and upper extremities, and female patients having more lesions located in the scalp, upper and lower back, and lower extremities. The BCCs in BCNS may range in number from one to well over a thousand. In addition, patients with BCNS may develop skin lesions that appear similar to nevi or seborrheic keratoses and tend to follow a more benign course, although after puberty the cutaneous lesions of BCNS tend to be more aggressive. The BCC lesions appear as flesh-colored to pink or tan dome-shaped papules that measure 1 to 10 mm ( Fig. 9-39, A ). Secondary changes such as ulceration, crusting, and bleeding rarely occur before puberty, but if left untreated these lesions can become extremely destructive. Unlike ordinary BCCs, BCNS-associated lesions do not appear to be induced by prolonged exposure to sunlight.
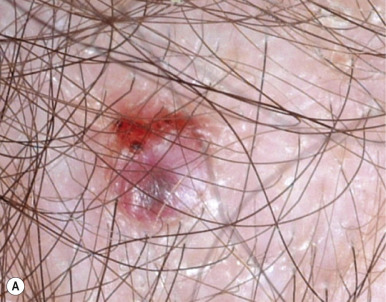
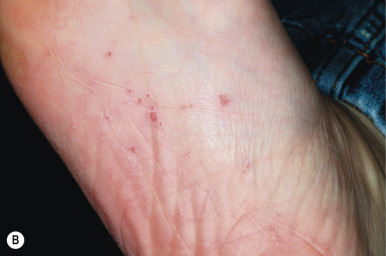
In addition to nevoid BCCs, affected individuals have a characteristic facies with coarse features, broad nasal root, hypertelorism, and other cutaneous stigmata. These include multiple small facial milia, comedones, large epidermal cysts, lipomas, fibromas, and café-au-lait macules. Shallow 2- to 3-mm palmar and plantar pits ( Fig. 9-39, B ), a characteristic feature of the syndrome, are seen in 70% to 90% of affected individuals. They may become more prominent after immersion of the hands or feet in water. These defective areas of keratinization usually first appear during the second decade of life or later. Palmar and plantar pits tend to have associated erythema, which on casual observation appears as multiple small red spots on the palms and soles.
Odontogenic jaw cysts occur in around 75% of patients and may present with painless swelling, jaw pain, abnormal taste, or a discharge in the mouth. The first jaw cyst often occurs before the age of 20 years, and symptoms referable to these lesions are often the presenting complaint. Jaw cysts occurring in patients with BCNS present earlier in life than those occurring in patients without BCNS. The cysts may be multiple and may result in loosening and loss of teeth. Patients with BCNS may become edentulous at an early age.
Musculoskeletal anomalies present in 60% to 80% of patients and include macrocephaly, frontoparietal bossing, high-arched palate, and broad nasal bridge. Other abnormalities include splayed or bifid ribs, mandibular and maxillary bone cysts, prognathism, kyphoscoliosis, Sprengel deformity, cervical or thoracic vertebral anomalies, spina bifida, pectus excavatum and carinatum, and shortened fourth metacarpals.
The most common neurologic abnormality is calcification of the falx cerebri, which is seen in up to 90% of patients with BCNS. In addition, mental retardation, electroencephalographic abnormalities, agenesis of the corpus callosum, seizures, hydrocephalus, and deafness may occur. Other associations include anosmia, renal malformations, endocrinopathy, and blindness. Patients with BCNS appear to have a predisposition to malignancy, especially medulloblastoma, which may present early in life. The desmoplastic subtype of medulloblastoma in children younger than 2 years of age is considered by some a major diagnostic criterion for the diagnosis of BCNS. This desmoplastic variant has a favorable prognosis. Ovarian and cardiac fibromas and meningiomas also occur at an increased rate in these patients.
The molecular cause of BCNS has been elucidated. Mutations in the human patched gene ( PTC ) have been identified in patients with BCNS as well as in those with sporadic BCCs. PTC is located on chromosome 9q and encodes a transmembrane receptor that represses growth-factor gene transcription. The gene product of PTC functions as a tumor suppressor, and PTC mutations results in dysregulation of several genes known to play a role in both organogenesis and carcinogenesis. Sequence analysis to detect mutations as well as deletion testing are both clinically available to assist in diagnosis of BCNS.
The management of patients with BCNS must be multidisciplinary and individualized. Genetic counseling is important given the autosomal dominant mode of inheritance. Family members judged to be at risk may be screened with skeletal surveys, dental radiographs, and neurologic evaluation (both clinical and radiographic). Molecular diagnosis is also possible (GeneDx, Gaithersburg, MD), with a sensitivity around 60% to 75%. Removal of cutaneous BCC may be accomplished with the same techniques as discussed earlier, and general anesthesia may be required for treatment of multiple lesions. Systemic retinoids (i.e., isotretinoin) may be useful in preventing BCCs, but its use must be balanced by potential side effects and in females the known teratogenicity. In addition, if the retinoid is discontinued, the patient may show rapid disease progression. Vismodegib, an inhibitor of the hedgehog signaling pathway approved for treatment of locally advanced or metastatic BCC, has been shown to reduce the BCC tumor burden and block growth of new lesions in patients with BCNS, though its potential benefit has been limited by adverse events that led to discontinuation by more than 50% of patients in recent study.
It is recommended that all pediatric patients with BCNS be monitored by a medical geneticist who ensures all appropriate referrals and follow-up evaluations. Patients require regular follow-up examinations with a dermatologist, pediatric dentist/oral surgeon, and ophthalmologist, as well as referral to a neurologist and psychologist as indicated. Annual vision, hearing, and speech screenings; routine developmental screening at well-child visits; and baseline brain MRI, digital Panorex of the jaw, and spinal films are also recommended.
Squamous Cell Carcinoma
Squamous cell carcinoma (SCC) is a malignant tumor of the epidermis rarely seen in children. Occasionally it may arise in normal skin, but generally it is seen in skin that has been injured by sunlight, trauma, thermal burn, or chronic inflammation. Children who develop SCC often have an underlying predisposing condition, including xeroderma pigmentosum (see Chapter 19 ), human papillomavirus infection (especially in the immunocompromised host), or a history of organ transplantation, chemotherapy with immunosuppression, or radiation therapy. In addition, scars related to dystrophic epidermolysis bullosa (see Chapter 13 ) may be predisposed to the development of SCC. Although these tumors generally do not present until the third or fourth decade, they may occasionally occur during childhood. SCC has also been reported to occur within the lesions of pansclerotic morphea of childhood.
The most common sites for SCC are the face (in particular the lower lip and pinna of the ear) and the dorsal aspect of the hands and forearms. Lesions usually present as red, scaly papules or plaques, often with induration. Telangiectasias may be present, as may ulceration or necrosis. Lesions that arise de novo usually appear as solitary, slowly enlarging, firm nodules with central crusting, underlying ulceration, and an indurated base. The histologic evaluation of SCC offers prognostic information based on the depth of the lesion and the cytologic features and degree of differentiation of the cells.
The prognosis for SCC of the skin is quite variable. Easily cured small lesions arising in sun-damaged skin have a low propensity to metastasize. Lesions arising in burn scars, prior radiation fields, and chronic wounds (i.e., ulcers or epidermolysis bullosa scars; see Chapter 13 ) tend to be more aggressive with higher rates of metastases. Complete excision is the treatment of choice for SCC. Other treatment options that may be considered include electrodesiccation and curettage, cryotherapy, photodynamic therapy, and MMS. Sun protection and skin self-examination education is again vital.
Keratoacanthoma
Keratoacanthoma (KA) is an epithelial tumor that may be clinically and histologically indistinguishable from SCC. These lesions are considered by many to be benign, and they often involute spontaneously without therapy. However, given rare reports of extensive local destruction and metastases, some consider KAs to be a variant of SCC. KAs are usually seen in older adults with rare reports of neonatal or childhood involvement. Multiple KAs characteristically have their onset in adolescence or early adult life and may occur in several clinical settings such as Ferguson Smith, Muir–Torre, or Witten–Zak syndromes. Generalized eruptive KA with the sudden onset of multiple lesions may also occur (termed the Grzybowski type ).
KA presents as a firm, dome-shaped nodule that generally measures 1 to 3 cm or more. The center contains a horny plug or is covered by a crust that conceals a central keratin-filled crater. The nodule generally grows rapidly and reaches its full size within 2 to 8 weeks. After a period of quiescence that may last for 2 to 8 weeks, most lesions heal spontaneously over several months with only a slightly depressed, somewhat cribriform scar in the previously affected area.
The main problem in the diagnosis of KA is its differentiation from SCC. In most cases, the rapid evolution and typical clinical appearance help to establish the correct diagnosis. Because the architecture of the lesion is as important as the cellular characteristics, full excisional biopsy is recommended to enable appropriate histopathologic evaluation. The treatment of KA is usually approached with a view toward its spontaneous resolution. Its close resemblance to SCC, however, often results in excisional biopsy. Other reported treatment options include intralesional corticosteroids, topical imiquimod, intralesional methotrexate, topical 5-fluorouracil, electrodesiccation and curettage, and radiation therapy.
Tumors of the Oral Mucosa
White Sponge Nevus
White sponge nevus is a rare, autosomal dominant condition that most often affects the oral mucosa and less commonly the mucosae of other regions. It presents as exuberant, asymptomatic white plaques on the buccal mucosae ( Fig. 9-40 ), palate, gingivae, or sides of the tongue. Other regions that may be involved include the nasal, vaginal, labial, and anal mucosae. In some instances, the plaques may be quite thickened with fissures and folds, and occasionally a raw, denuded surface may be evident. White sponge nevus may be present at birth or may have its onset any time from infancy through adolescence. The lesions are most commonly misdiagnosed as candidiasis in young children. It reaches its maximum severity during adolescence or early adulthood, and the lesions are entirely asymptomatic and often noted incidentally during examination. Both genders are equally affected. The differential diagnosis of white sponge nevus may include oral leukoplakia, pachyonychia congenita, dyskeratosis congenita, cheek biting, chemical burn, syphilis, betel nut or tobacco use, and lichen planus.
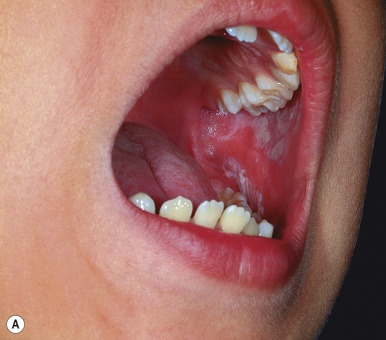
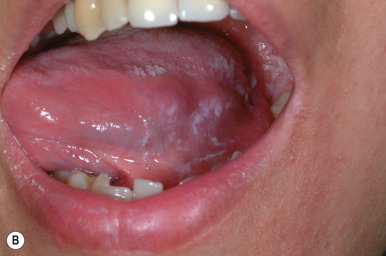
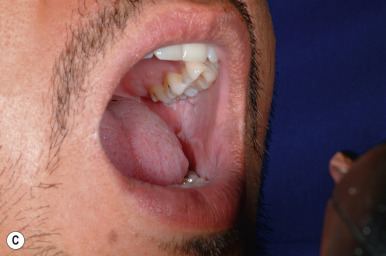
Defects in the genes for keratins 4 and 13 have been demonstrated to be responsible for white sponge nevus. This disorder is generally benign and asymptomatic and requires no specific therapy. White sponge nevus should not be confused with focal epithelial hyperplasia (Heck disease), a rare disorder of the oral mucosa of children believed to be caused by infection with human papillomavirus, especially types 13 and 32. In patients with Heck disease, the mucosa of the lower lip is most commonly involved and reveals soft papules and plaques that tend to regress spontaneously over several months.
Leukoplakia
Leukoplakia is a term generally used to describe a white plaque involving the oral mucosa that cannot be easily removed by scraping or rubbing with a cotton swab or tongue blade. It is a nonspecific term inconsistently applied to a variety of different etiologic lesions, including white sponge nevus, pachyonychia congenita, dyskeratosis congenita, hereditary benign intraepithelial dyskeratosis, hereditary mucoepithelial dysplasia, and many others. It is also used to describe white changes on the vulva of females, where its use is equally nonspecific.
Oral leukoplakia is the terminology usually used to describe a condition seen most often in adults and is related to a variety of factors including trauma, poor oral hygiene, chronic irritation, and use of tobacco products. It may occasionally be seen in children and has been reported in pediatric patients with HIV infection. The clinical presentation is notable for either focal or more diffuse involvement with white plaques of the buccal mucosae, hard or soft palate, lateral surfaces of the tongue, and the floor of the mouth. Mucosal biopsy with histopathologic evaluation is usually necessary to confirm the exact diagnosis, and treatment depends on the etiology. Meticulous attention to oral hygiene is useful regardless of the cause. Since oral leukoplakia may have malignant potential (eventuating into SCC), long-term follow-up observation is indicated, with repeat tissue biopsy when necessary.
Tumors of the Epidermal Appendages
Nevus Sebaceous
Nevus sebaceous of Jadassohn is a common congenital lesion that occurs mainly on the face and scalp. These lesions are usually solitary and present as a well-circumscribed, hairless plaque. They are generally present at birth but may first be noted during early childhood and rarely in adult life. Although rare familial forms have been reported, these hamartomas tend to be sporadic. Multiple nevi sebaceous may occur in association with cerebral, ocular, and skeletal abnormalities as part of an ENS. This association has been termed Schimmelpenning syndrome. Recently, somatic mutations in HRAS and KRAS were demonstrated in lesions of nevus sebaceous.
Classic nevus sebaceous presents as a hairless, yellow to tan plaque on the scalp or face ( Figs. 9-41 and 9-42 ). The surface may be verrucous or velvety, and the lesions are often oval or linear. In some patients hyperpigmentation may be a prominent feature, making the distinction from a verrucous epidermal nevus difficult. Nevus sebaceous may vary in size from a few millimeters to several centimeters in length. Their yellow color is related to sebaceous gland secretion, and often this color becomes less prominent after infancy. The lesions tend to enlarge proportionate with growth of the child, until puberty when they may become significantly thicker, more verrucous, and more greasy in appearance ( Fig. 9-43 ) as a result of hormonal stimulation of the sebaceous glands within them. Papillomatous projections may occur, sometimes simulating the appearance of verruca vulgaris.