The inborn disorders of metabolism are a group of primarily autosomal recessive hereditary disorders that result in metabolic and clinical defects. (More comprehensive discussion of some of these topics is covered elsewhere: disorders of tyrosine metabolism [Richner–Hanhart syndrome], Gaucher syndrome, and multiple sulfatase deficiency are mentioned in Chapter 5 ; Menkes syndrome in Chapter 7 ; alkaptonuria in Chapter 11 ; Fabry disease and fucosidosis in Chapter 12 ; and Hartnup disease in Chapter 19 .) Most developed countries have instituted comprehensive screening programs during the first week of life that allow detection of many of these defects, among them biotinidase deficiency, aminoacidopathies, urea cycle defects, and organic acid disorders.
Phenylketonuria
Phenylketonuria (PKU; phenylpyruvic oligophrenia) is an autosomal recessive disorder of amino acid metabolism characterized by mental retardation, diffuse hypopigmentation, seizures, dermatitis, and photosensitivity ( Table 24-1 ). The classic form is caused by a deficiency of phenylalanine hydroxylase or its cofactor, tetrahydrobiopterin. Its incidence has been estimated to be 1 in 10,000 births. Because of mandatory screening in the neonatal period, the disorder is detected early in developed countries, and the clinical features do not appear if early dietary control is achieved. Screening is achieved by a variety of different tests ranging from a semiquantitative bacterial inhibition assay (Guthrie test) to mass spectrometry assays. If missed in the neonatal period by screening, the diagnosis depends on the demonstration of elevated serum levels of phenylalanine (20 mg/dL or higher, 10 to 50 times that of normal), normal or elevated levels of plasma tyrosine (normal is approximately 1 mg/dL), or elevated urinary levels of phenylpyruvic acid. The latter can be detected by a characteristic green or blue color that results when a few drops of urine are added to a 10% solution of ferric chloride. The normal phenylalanine/tyrosine ratio is 1 : 1; in PKU it is greater than 3 : 1. The observed pigmentary dilution is thought to result from the inhibitory effect of phenylalanine on tyrosinase.
| Disorder | Inheritance | Defect | Clinical Features | Diagnosis | Treatment |
|---|---|---|---|---|---|
| Phenylketonuria | AR | Phenylalanine hydroxylase | 90% are blond, blue-eyed, and fair-skinned; musty odor; sclerodermatous plaques; eczematous dermatitis; photosensitivity; retardation; microcephaly; short stature | Guthrie test, ferric chloride test | Low phenylalanine diet; sapropterin dihydrochloride |
| Homocystinuria | AR | Cystathionine synthetase | Ectopia lentis, myopia, arachnodactyly, seizures, mental retardation, cerebrovascular accidents, sparse light or blond easily friable hair, malar flush, wide-pored facies, livedoid rash, “Charlie Chaplin-like” gait and “rocker-bottom” feet | Cyanide nitroprusside test | Low methionine diet with pyridoxine (vitamin B 6 ) |
| Alkaptonuria (ochronosis) | AR | Homogentisic acid oxidase | Dark urine; blue to brownish-black pigment on nose, malar region, sclerae, ears, axillae, genitalia; staining of clothing (“beads of ink” perspiration); arthritis; contractures; rupture of Achilles tendon; mitral and aortic valvulitis and/or calcific aortic stenosis | Ferric cyanide reduction | Nitisinone |
| Trimethylaminuria | AR | Flavin mono-oxygenase type 3 | Odor of rotting fish | Trimethylamine in urine | Dietary avoidance of choline |
| Wilson disease (hepatolenticular degeneration) | AR | ATP7B ; ceruloplasmin metabolism | Kayser–Fleischer rings, hyperpigmentation, azure lunulae, neurologic and liver dysfunction | Aminoaciduria, decreased ceruloplasmin | Chelating agents (BAL, versenate, or D-penicillamine; trientine has fewer side effects, but is weaker and more expensive); zinc to sequester copper |
| Lesch–Nyhan syndrome | X-LR | HPRT deficiency | Mental retardation, spastic cerebral palsy, choreoathetosis, self-mutilation | Increased uric acid, orange uric acid crystals in diaper | Allopurinol |
Most commonly seen in individuals of northern European ancestry, 90% of affected individuals are blond, blue-eyed, and fair-skinned, given the important role of phenylalanine and its derivative tyrosine in pigment formation. A peculiar musty odor, attributable to decomposition products (phenylacetic acid or phenylacetaldehyde) in the urine and sweat, and early intractable vomiting are characteristic. Infants appear to be normal at birth and if untreated, develop manifestations of delayed intellectual development sometime between 4 and 24 months of age. Seizures, attention-deficit/hyperactivity disorder, hypertonicity, hyperreflexia, a peculiar gait, speech delay, and self-destructive behavior have been described. Skeletal changes include microcephaly, short stature, pes planus, and syndactyly. The dermatitis appears in 10% to 50% of untreated patients and resembles atopic dermatitis; sclerodermoid changes of the proximal extremities, sparing the hands and feet, may present during the first 2 years of life. These changes regress with appropriate treatment.
Management consists of a diet low in phenylalanine content, starting at as early an age as possible. This can be initiated by the use of formula from which most of the phenylalanine has been removed or a synthetic amino acid preparation devoid of phenylalanine. Restriction of phenylalanine leads to reversal of the seizures, dermatitis, and pigmentary dilution, but the effect on intellectual function depends on the age at which therapy is initiated. Little benefit for intellectual function can be achieved if therapy is initiated after 2.5 years of age, and optimal benefit is achieved if it is initiated by 2 months of age. It should be remembered that phenylalanine is high in many foods and is a metabolite of the sweetener aspartame. Children and adolescents with PKU who achieve dietary control early in life are well adjusted and have an excellent quality of life. Sapropterin dihydrochloride may reduce phenylalanine levels in patients who do not adequately adhere to the prescribed diet and has been shown to significantly improve within 4 weeks the attention-deficit hyperactivity disorder and executive function of patients.
Although there is some controversy about continuation of phenylalanine restriction in later childhood and adults, lack of dietary control during the 1st trimester of pregnancy of affected mothers (>600 µM or 10 mg/dL phenylalanine) has led to cardiac defects in 15% of offspring, particularly coarctation of the aorta and hypoplastic left heart. Optimal levels during pregnancy of 120 to 360 µM/L are recommended. Some patients maintain low levels of phenylalanine as adults without dietary manipulation.
Disorders of Tyrosine Metabolism
Clinical disorders of tyrosine metabolism include neonatal tyrosinemia, tyrosinemia I, and tyrosinemia II (Richner–Hanhart syndrome). Of these, only Richner–Hanhart syndrome exhibits cutaneous manifestations and is described in Chapter 5 . These include palmoplantar keratoderma with painful erosions and photophobia with corneal erosions.
Cobalamin Deficiency
In a single patient, a metabolic disorder of vitamin B 12 (cobalamin), called cblJ, resulted from biallelic mutations in ABCD4 , which encodes an ABC transporter involved in the intracellular processing of cobalamin. Features were prominent, diffuse progressive skin hyperpigmentation, but hair lightening, with macrocytic anemia and cobalamin deficiency. Treatment with oral cobalamin (3 mg daily for this 12-year-old) led to metabolic correction and some reduction in the skin hyperpigmentation.
Homocystinuria
Homocystinuria is an autosomal recessive disorder of methionine metabolism that results from an absence or deficiency of cystathionine β-synthase, the pyridoxine-dependent hepatic enzyme that catalyzes the formation of cystathionine from homocystine and serine. Based on biochemical testing, the disorder is most common in Ireland (1 in 65,000 births) and has a worldwide incidence of 1 in 344,000. However, a study in Norway using genotyping showed an estimated birth prevalence of 1 in 6400.
The disorder affects predominantly the eyes, central nervous system, blood vessels, and bones (see Table 24-1 ). Subluxation of the ocular lenses (ectopia lentis) occurs in 82% of patients by 15 years of age and is usually present by 10 years of age. Developmental delay is evident during infancy and most untreated patients develop seizures and significant retardation. Bony abnormalities include a body habitus similar to that seen in Marfan syndrome (see Chapter 6 ), and generalized osteoporosis, which is seen radiographically in 50% of affected individuals by 15 years of age. Platyspondylia (congenital flattening of the vertebral bodies) and hollowing out of the vertebral bodies by pressure of the vertebral disks are also seen radiographically, and kyphoscoliosis, pectus carinatum, and genu valgum are common. Some patients have rocker-bottom feet and most have a shuffling toe-out “Charlie Chaplin-like” gait. Megaloblastic anemia has also been described.
The vascular complications can be life-threatening; the chance of a vascular event during childhood is 25% to 30% and increases to 50% by 30 years of age. These include pulmonary embolism, myocardial infarction, transient ischemic attacks, cerebrovascular accidents, abdominal aortic aneurysm, and venous thromboses. Cutaneous features include sparse, light or blond, coarse hair that can darken and become softer with treatment; a malar flush; coarse, wide pores on facial skin; and a reticulated livedoid vasculopathy (cutis reticulata) on the face and extremities.
The presence of homocystinuria is suggested by the clinical features and can be confirmed by a urinary cyanide or sodium nitroprusside testing (turns a beet-red color) and by amino acid chromatography of the serum and urine. The presence of homocystine in the urine establishes the diagnosis. In the blood, homocysteine and methionine are elevated, whereas cysteine levels are decreased. Unfortunately, there are no neonatal manifestations of homocystinuria, and not all newborns with this disorder have increased blood methionine levels.
In at least 50% of patients, the disease is responsive to pyridoxine and can be controlled with the combination of pyridoxine (vitamin B 6 : 150 mg/day for an infant to 500 mg/day for an adolescent), folic acid, and vitamin B 12 (cobalamin). For those who do not respond to vitamins alone, a methionine-restricted, cystine-supplemented diet is required as well. Betaine, a methyl donor that remethylates homocysteine to methionine, has been used as an adjunct to treatment, especially in patients who do not respond to vitamin therapy. As with PKU, early institution of therapy dramatically reduces the risk of complications.
Trimethylaminuria
Trimethylaminuria (the fish odor syndrome) is a metabolic disorder in which accumulation of trimethylamine in the sweat and urine gives rise to an unpleasant “rotting fish” odor to the skin, sweat, and urine. There are no visible cutaneous changes, but one-third of patients with an unexplained intermittent malodor were shown to be positive for the disorder using a standard choline challenge.
The disorder results from mutations in the flavin-containing monooxygenase type 3 ( FM03 ) gene. The liver is unable to convert the trimethylamine generated in the intestinal tract by bacterial degradation of choline- and lecithin-containing foods such as saltwater fish and seafood, egg yolk, liver, kidney, soy beans, meat, broccoli, cauliflower, cabbage, Brussel sprouts, wheat germ, milk (from wheat-fed cows), and yeast to the nonodorous oxide metabolite. The psychosocial effects of the condition can be devastating, including disruption of schooling, clinical depression, and attempted suicide. Treatment is best accomplished by avoidance and limitation of choline-containing foods, although administration of metronidazole has also led to a clinical and biochemical response.
Wilson Disease (Hepatolenticular Degeneration)
Wilson disease is an autosomal recessive disorder that results from mutations in ATP7B , which encodes a copper-transporting adenosinetriphosphatase (ATPase). Deficiency leads to an inability to synthesize normal amounts of ceruloplasmin. Patients show abnormal hepatic excretion of copper and toxic accumulations of copper in the liver, brain, and other organs. The clinical triad that results consists of progressive neurologic dysfunction, hepatic cirrhosis, and pathognomonic pigmentation of the corneal margins (Kayser–Fleischer ring) (see Table 24-1 ).
The disorder can be detected as early as 4 years of age but usually manifests during adolescence. The clinical signs at presentation can be quite varied. Although initial signs of the disorder are often neurologic (intention tremors, dysarthria, ataxia, incoordination, personality changes, psychiatric problems, and worsening school performance and handwriting), signs of liver insufficiency (jaundice, ascites, hepatomegaly, hematemesis, and cutaneous spider angiomas) are present at onset in 40% to 50% of affected individuals.
Hyperpigmentation of the anterior lower legs, genital region, or even in a generalized distribution may be the presenting sign and may be misinterpreted as Addison disease. In addition, patients may manifest a vague greenish discoloration on the skin of the face, neck, and genitalia. Increased melanin deposition, but not copper or iron, is seen in cutaneous biopsy specimens. Blue or azure lunulae of the nails have been reported in individuals with this disorder, but also have been observed in normal individuals as a complication of phenolphthalein or quinacrine ingestion, and in argyria. White discoloration of the nails has also been noted. Xerosis has been described in 45.7% of affected children.
Kayser–Fleischer rings are seen as dense golden brown or greenish brown pigmentation localized near the limbus of the cornea. Occurring in about 95% of patients with this disorder and best visualized by side lighting and slit-lamp examination, this discoloration is produced by the deposition of copper in the Descemet membrane at the periphery of the cornea. Kayser–Fleischer rings may also be seen in other copper overload states such as primary cirrhosis, intrahepatic cholestasis of childhood (Byler syndrome), and chronic active hepatitis.
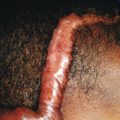
Hepatolenticular degeneration is a progressive disease, and if untreated, is inevitably fatal. Death usually results from infection, hepatic disease, or liver failure. Treatment depends on removal of accumulated copper depositions in the body. Chelating agents such as D-penicillamine or trientine (triethylene tetramine dihydrochloride) and elemental zinc (1 to 3 mg/kg per day divided three times daily) are helpful. Because discontinuation of therapy can result in rapid deterioration, treatment is life long. Penicillamine can produce allergic or toxic reactions, fever, cutaneous rashes, leukopenia, thrombocytopenia, elastosis perforans serpiginosa (see Chapter 6 ; Fig. 6-13 ), other dermal disorders, and immunobullous skin disease. Because penicillamine inhibits pyridoxine-dependent enzymes, patients should also be given daily supplements of pyridoxine (12.5 to 50 mg/day). In an effort to minimize copper deposition, patients should be encouraged to limit their ingestion of copper-rich foods such as liver, nuts, chocolate, cocoa, mushrooms, brain, shellfish, dried foods, and broccoli. Liver transplantation can be life saving for patients with hepatic failure.
Lesch–Nyhan Syndrome
Lesch–Nyhan syndrome, an X-linked recessive disorder of purine metabolism, is characterized by mental retardation, choreoathetoid movements, and self-mutilation. The condition is caused by a deficiency of hypoxanthine-guanine phosphoribosyl transferase (HPRT), which leads to an overproduction of uric acid and the clinical features associated with this disorder ( Table 24-1 ). Patients appear normal at birth and may develop normally for 6 to 8 months. The first recognizable sign of the disease is often orange uric acid crystals (resembling grains of sand) in the diaper or as hematuria during the early months of life.
The onset of cerebral manifestations may be subtle, with difficulty in sitting or standing without help, involuntary movements, dystonia, spasticity, and increased deep tendon reflexes. Although mental retardation may vary in degree, it usually is severe, and abnormal behavior remains a striking characteristic of the disease. The main clinical feature of this disorder is a loss of tissue about the mouth or fingers that occurs, not because of an inability to feel pain, but as a result of the child’s habit of compulsive self-destructive biting of these areas. In time, without adequate restraints, all of the lower lip accessible to the teeth may be chewed away. The face, fingers, and wrists may also be mutilated, and because young children commonly bite others, caution should be exercised when handling children with this disorder. Other destructive behaviors include head banging, extension of arms when being wheeled through doorways, tipping of wheelchairs, eye-poking, fingers in wheelchair spokes, and rubbing behaviors. Other manifestations of hyperuricemia, such as tophaceous deposits, nephropathy, and gouty arthritis, occur later. Reticulated pigment changes in a variant form have been described.
Not to be confused with familial dysautonomia (Riley–Day syndrome) and congenital insensitivity to pain with anhidrosis (see Chapter 8 ), other conditions characterized by biting and self-mutilating behavior, Lesch–Nyhan syndrome can be confirmed by the clinical presentation and laboratory demonstration of increased levels of uric acid in the blood and urine. Heterozygous females are asymptomatic. Management includes the use of allopurinol (in dosages of 10 mg/kg per day or, if >10 years of age, 600 to 800 mg/day divided twice daily) in an effort to control uric acid levels. Physical restraints (hand bandages and elbow splints) may be used to help control the self-mutilating behavior of patients. Lip biting may require extraction of deciduous teeth, but permanent teeth should be spared, because lip biting usually diminishes with age.
Dermatitis From Nutritional Abnormalities
Acrodermatitis Enteropathica
Acrodermatitis enteropathica is an autosomal recessive disorder that generally appears in early infancy and is characterized by a triad of acral and periorificial skin lesions (vesiculobullous, pustular, and eczematous), diarrhea, and alopecia ( Table 24-2 ). The condition first becomes manifest days to weeks after birth if an infant is bottle-fed and shortly after weaning if breastfed. The disorder results from mutations in SLC39A , which encodes an intestinal zinc transporter ZIP4.
| Disorder | Cause | Features | Evaluation | Therapy |
|---|---|---|---|---|
| Zinc deficiency | Acrodermatitis enteropathica Decreased zinc in breast milk Malabsorption Zinc-poor IV hyperalimentation | Periorificial dermatitis, stomatitis, glossitis, alopecia, irritability, diarrhea, failure to thrive, candidal infection, photophobia | Zinc levels, serum alkaline phosphatase; zinc levels in maternal breast milk | Zinc supplementation |
| Biotin deficiency | Holocarboxylase deficiency Biotinidase deficiency Malabsorption Ingestion of egg whites Biotin-poor IV hyperalimentation | Periorificial dermatitis, conjunctivitis, blepharitis, alopecia, candidal infections, metabolic acidosis, vomiting, lethargy, developmental delay, seizures, ataxia, optic atrophy, hearing loss | Urinary organic acids, plasma ammonia and lactate, serum biotin, specific enzyme activity | Biotin supplementation |
| Deficiency of vitamin B 12 or isoleucine from restrictive diets | Organic acidurias Citrullinemia Maple syrup urine disease Methylmalonic acidemia Propionic acidemia | Periorificial dermatitis, alopecia, candidal infections, metabolic acidosis, vomiting, failure to thrive, lethargy, developmental delay, hypotonia, pancytopenia | Urinary organic acids; plasma ammonia and glycine levels, plasma isoleucine levels | Dietary manipulation (results from effects of low-protein diet; some respond to vitamin B 12 supplementation) |
| Cystic fibrosis | Deficiency of CFTR | Periorificial and truncal dermatitis, occasionally alopecia, failure to thrive, diarrhea, irritability, edema | Sweat test, serum albumin, gene analysis, mutational analysis of CFTR gene | Pancreatic enzymes, vitamin and zinc supplementation |
| Essential fatty acid deficiency | Inadequate dietary intake | Generalized scaling with underlying erythema, intertriginous erosions, alopecia, decreased pigmentation of hair, failure to thrive, thrombocytopenia, and anemia | Plasma linoleic, linolenic, arachidonic and icosatrienoic acid levels; check for other deficiencies | Supplementation of essential fatty acids |
| Kwashiorkor | Inadequate protein; often infants taken off milk protein for allergies or dietary fads without adequate replacement | Superficial desquamation with erosion (flaky paint), edema with moon facies and pallor, hyperpigmentation and hypopigmentation, ecchymoses, sparse, dry lusterless hair with decreased pigmentation and “flag sign”, cheilitis, dry mucosae, irritability and failure to thrive, irritability and apathy, secondary skin infections, diarrhea, hepatomegaly | Serum albumin, good dietary history | Slow supplementation with protein, correction of electrolyte imbalances |
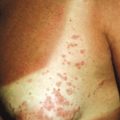
The basic cutaneous lesion of acrodermatitis enteropathica is an erythematous, scaling, crusted, psoriasiform, eczematous, or vesiculobullous eruption (see Fig. 2-29 ). Lesions tend to be localized around the body orifices (mouth, nose, ears, eyes, and perineum; Fig. 24-1 ) and symmetrically located on the buttocks ( Fig. 24-2 ) and extensor surface of major joints (elbows, knees, hands, and feet), the scalp, and the fingers and toes (hence the term acrodermatitis ). On the face, the eroded and crusted peribuccal plaques may appear impetiginized, and secondary infection with Candida albicans is common. When the fingers and toes are involved, there is marked erythema and swelling of the paronychial tissues, often with subsequent nail deformity. If unrecognized or untreated, acrodermatitis enteropathica follows an intermittent but relentlessly progressive course, and as a consequence of general disability, infection, or both, commonly ends in death.
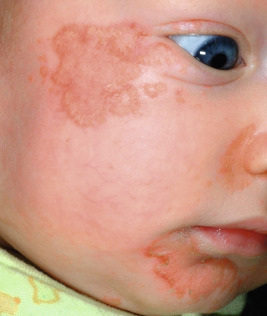
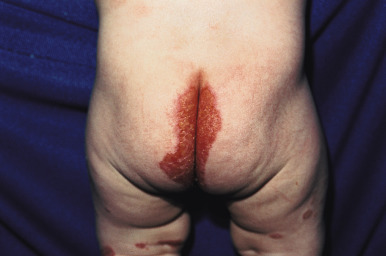
Typically, infants with acrodermatitis enteropathica are listless, anorexic, and apathetic. Many infants show frequent crying, irritability, and restlessness. Tissue wasting is present with an associated failure to thrive. During periods of exacerbation, frothy, bulky, foul-smelling diarrheal stools are present. Other findings include conjunctivitis, photophobia, stomatitis, perlèche, nail dystrophy, recurring candidal or bacterial infection, and alopecia of the scalp, eyelashes, and/or eyebrows. Children suffering from this disorder exhibit a striking uniformity of appearance, mainly because of the alopecia and periorificial lesions.
In addition to the classic inherited disease, zinc deficiency with signs mimicking acrodermatitis enteropathica may also occur in babies who are breastfed by mothers with inadequate secretion of zinc in the maternal milk. Maternal mutations in SLC30A2 ( ZnT-2 ), which encodes a protein required for zinc secretion, have been found. Zinc deficiency with clinical manifestations may also be seen in individuals receiving long-term parenteral nutrition with inadequate zinc supplementation, patients who have had intestinal bypass procedures, premature infants with low zinc storage (particularly those fed exclusively with human milk), in patients with Crohn disease, in infants with human immunodeficiency virus (HIV) infection and diarrhea (causing inadequate absorption of zinc), in patients with cystic fibrosis with poor zinc absorption, patients with celiac disease, chronic alcoholics or patients with anorexia nervosa, in individuals on zinc-deficient vegetarian diets, and in patients with low pancreatic enzyme levels resulting in poor intestinal absorption of zinc. In babies with cystic fibrosis, the manifestations of zinc deficiency typically are noted at 3 to 5 months of age, before any evidence of pulmonary disease. The low serum albumin level and abnormal sweat test can confirm the diagnosis. An acrodermatitis enteropathica-like eruption can be seen with biotin deficiency and kwashiorkor.
The diagnosis of acrodermatitis enteropathica is based on the clinical features and low serum zinc levels (50 mcg/dL or lower). It should be noted, however, that zinc contamination of glass tubes and rubber stoppers often occurs; blood samples should be collected in acid-washed sterile plastic tubes with the use of acid-washed plastic syringes. Low serum alkaline phosphatase levels (even when the serum zinc level is normal), low serum lipid levels, and defective chemotaxis may be found. Skin biopsy is not diagnostic but may be useful in some patients. Zinc levels can also be measured in maternal milk.
Zinc sulfate supplementation is provided at 5 to 10 mg/kg per day, which usually provides approximately 1 to 2 mg/kg per day elemental zinc, although the dosage may differ to provide sufficient elemental zinc if other formulations are used. When given in divided doses two or three times a day, improvement in temperament and decrease in irritability can generally be noted within 1 or 2 days; the appetite improves in a few days, and diarrhea and skin lesions begin to respond within 2 or 3 days after the initiation of therapy. Hair growth begins after 2 to 3 weeks of therapy, and increase in the growth of the infant generally occurs within approximately 2 weeks. After the patient’s condition appears to be stabilized, zinc levels should be monitored at periodic (6-month) intervals, followed by the adjustment of supplemental zinc to the lowest effective dosage schedule. Because foods may have an effect on the absorption of zinc, zinc supplementation should be administered 1 or 2 hours before meals. Zinc supplementation can cause gastrointestinal upset (nausea, vomiting, gastric hemorrhage); zinc gluconate tends to cause fewer gastrointestinal problems. Less zinc supplementation may be required for acquired or dietary zinc deficiency. In infants who have zinc deficiency as a result of defective maternal mammary zinc secretion, zinc supplementation can be discontinued after weaning.
Disorders That Resemble Acrodermatitis Enteropathica
Biotin Deficiency/Organic Acid Disorders
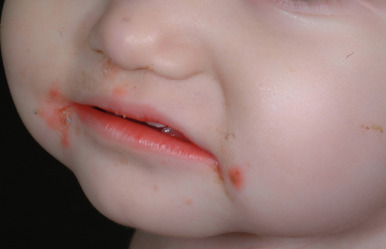
A dermatitis resembling acrodermatitis enteropathica, often in association with alopecia and changes in hair texture, has been described in patients with biotin deficiency and in certain organic acid disorders. The cutaneous features resembling acrodermatitis enteropathica have been described in methylmalonic acidemia, propionic acidemia, glutaric aciduria type I, maple syrup urine disease, ornithine transcarbamylase deficiency, and citrullinemia, because of dietary restriction and deficiency of amino acids, especially isoleucine ( Fig. 24-3 ). Acrodermatitis enteropathica-like eruptions have also been reported in children with Hartnup disease (see Chapter 19 ).
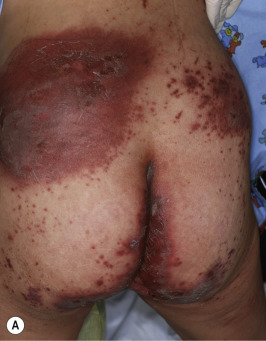
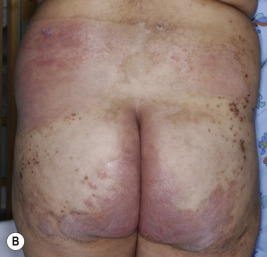
Biotin, part of the vitamin B complex, is required for the function of four carboxylase enzymes :
- 1.
3-Methyl crotonyl-CoA carboxylase, essential for the catabolism of leucine
- 2.
Propionyl CoA carboxylase, essential for the catabolism of isoleucine, threonine, valine, and methionine
- 3.
Pyruvic acid carboxylase, required for the gluconeogenesis and regulation of carbohydrate metabolism
- 4.
Acetyl CoA carboxylase, an enzyme of long-chain fatty acid synthesis that contains biotin.
Biotin deficiency may be induced by a biotin-deficient diet; that is, a diet in which patients ingest large quantities of raw egg white (which contains the protein avidin that binds to biotin, thus preventing its absorption in the intestine) and/or prolonged parenteral nutrition to which biotin has not been added (see Table 24-2 ). The resulting deficiency is manifested by anorexia, lassitude, a pale tongue, grayish pallor of the skin, atrophy of the lingual papillae, hair loss, anemia, muscle pains, dryness of the skin, and a scaly dermatitis, all of which disappear after biotin administration or by cooking, boiling, or steaming of egg white (which causes avidin to lose its biotin-binding capacity). Decreased biotin levels have also been noted in patients who have been administered antiepileptics (phenytoin, carbamazepine), and may result in seborrheic dermatitis and alopecia.
Several autosomal recessive disorders may manifest as an acrodermatitis enteropathica-like eruption, because they require biotin as a cofactor. Cheilitis and diffuse erythema with erosions and desquamation are features of methylmalonic acidemia before therapeutic amino acid restriction. Inherited biotin deficiency also occurs in two forms of multiple carboxylase deficiency, one of which presents acutely during the neonatal period (neonatal form), and the other that presents during early infancy (juvenile form). These disorders occur in up to 1 : 40,000 births. Both forms usually show a sharply marginated, brightly erythematous scaling eruption of the periorificial areas, scalp, eyebrows, and eyelashes ( Fig. 24-4 ). Alopecia can manifest as hair thinning or total alopecia. Secondary candidiasis is not uncommon. The neonatal form, which results from deficiency of holocarboxylase synthetase, appears in the first few weeks of life with metabolic acidosis, ketosis, and rarely, cutaneous manifestations. Patients show feeding and breathing difficulties, seizures, hypotonia, and lethargy; without early diagnosis and treatment, they usually die. The juvenile form of the disorder, which first manifests at 2 months of age or older, is the result of deficiency of biotinidase, which is required for recycling of endogenous biotin. Patients have low levels of biotin in the blood and urine and impaired biotin absorption and/or transport. In addition to the cutaneous features, the juvenile form is characterized by seizures (especially myoclonic spasms that respond partially to antiepileptics), hyperventilation or apnea, recurrent infections (including candidal skin infections), keratoconjunctivitis, ataxia, hypotonia, glossitis, and life-threatening acidosis and massive ketosis. More than 50% of patients whose disease is not recognized early have hearing loss that does not improve with biotin therapy.
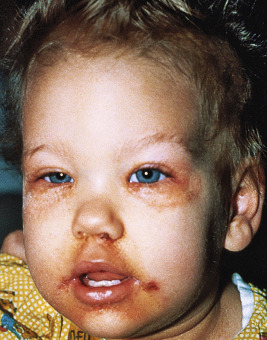
The diagnosis of biotin deficiency can be established by a decreased concentration of plasma biotin. Deficiencies of biotinidase or holocarboxylase synthetase also show an increase in the urinary metabolites 3-hydroxyisovaleric acid, 3-methylcrotonyl-glycine, 3-hydroxypropionic acid, methylcitric acid, and lactic acid. Biotin deficiency can be treated by intravenous multivitamins containing 60 mcg of biotin or by the oral administration of 5 to 10 mg of biotin daily. Higher concentrations of biotin may be required for holocarboxylase synthetase deficiency. Skin and neurologic signs tend to improve within a few weeks, although neurologic dysfunction can be permanent if treatment is delayed.
Kwashiorkor
Kwashiorkor is a severe form of protein deficiency in which edema, hypoalbuminemia, and dermatosis predominate. Seen primarily in underdeveloped countries, several cases of kwashiorkor have been described in developed countries as well. Cases in developed countries have been described because of nutritional ignorance, food allergen avoidance, food fads, and child abuse. Particularly common is the substitution of rice “milk,” which has only a miniscule content of protein and is not a milk substitute. In children with chronic protein and caloric malnutrition, particularly from Africa, signs usually present during the second or third year of life with the onset of weaning from breastfeeding. In contrast, patients in developed countries most commonly show features during the first year of life with a rapid onset of edema. Kwashiorkor has also been described with malabsorption disorders, including infantile Crohn disease and cystic fibrosis.
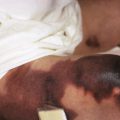
The clinical picture consists of a blanching erythema with an overlying reddish-brown scale that shows a sharply marginated raised edge. This edge resembles paint that is lifting up and about to peel off, leading to the term flaky paint dermatosis ( Figs. 24-5 and 24-6 ). In contrast to lesions of pellagra, the dermatosis seldom appears on areas exposed to sunlight and tends to spare the feet and dorsal areas of the hands. Photosensitivity, purpura, and excessive bruisability may also be present. Other associated features include changes in mental behavior, anorexia, apathy, irritability, growth retardation, and fatty infiltration of the liver with hypoproteinemia. As a result of the hypoalbuminemia, affected children show edema of the face (moon facies) ( Fig. 24-7 ), feet, and abdomen (potbelly appearance).
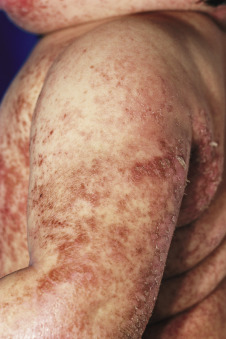
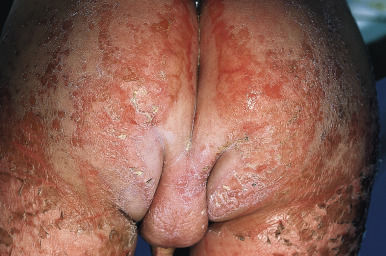
In mild cases the cutaneous eruption is associated with a superficial desquamation; in severe cases there are large areas of erosion that have been mistaken for Stevens–Johnson syndrome and/or toxic epidermal necrolysis. As the disease progresses, the entire cutaneous surface develops a reddish or coffee-colored hue. Other associated features include circumoral pallor, loss of pigmentation (especially after minor trauma), and depigmentation of the hair. In darker haired children, the hair color can change to a reddish-brown hue or even gray or straw color ( Fig. 24-8 ). The dyschromia with hypopigmentation has been attributed to deficiency of tyrosine, which is critical for melanin synthesis. When periods of malnutrition alternate with intervals of adequate dietary intake, alternating bands of light and dark color (the flag sign) are produced in the hair.
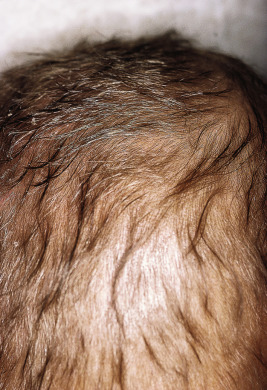
The condition may be difficult to distinguish from acrodermatitis enteropathica, and in fact, children may have concomitant zinc deficiency as well. The hypoalbuminemia and edema are helpful signs to distinguish kwashiorkor from acrodermatitis enteropathica and other disorders of nutritional deficiency. Children with marasmus have both protein and caloric deficiency. They appear emaciated (vs. the edematous appearance of children with kwashiorkor), have dry, scaling skin that may show follicular hyperkeratosis, and often have thin, sparse hair.
Children afflicted with kwashiorkor are extremely ill, and if they are not treated, the mortality rate can be 30% or more, primarily resulting from infection (impairment of immunity) and electrolyte imbalance with diarrhea. In underdeveloped countries where the disorder is common, breastfeeding should be continued for as long as possible to prevent the protein malnutrition. Treatment of affected individuals with gradual administration of a high-protein diet, vitamin supplementation, and correction of dehydration and electrolyte imbalance leads to resolution of the clinical abnormalities.
Essential Fatty Acid Deficiency
Essential fatty acid deficiency (EFA) can also be manifested by periorificial dermatitis and a generalized xerotic or eczematous dermatitis (see Table 24-2 ). Failure to thrive, alopecia with lightly colored hair, and thrombocytopenia are other signs that may occur. The condition usually occurs in patients receiving parenteral nutrition without lipid supplementation, in association with severe fat malabsorption from gastrointestinal disorders, or with surgery of the gastrointestinal tract. EFA has also been described in patients with cystic fibrosis, anorexia nervosa, and acrodermatitis enteropathica. Decreased plasma levels of linoleic, linolenic, arachidonic, and icosatrienoic acid, as well as an icosatrienoic/arachidonic acid ratio of greater than 0.4 confirms the diagnosis. The treatment consists of oral or parenteral fat emulsions. If these cannot be administered, topical application of 2 to 3 mg/kg soybean or safflower oil may be sufficient to restore plasma levels of linoleic acids but may not maintain stores in the liver or other tissues.
The Hyperlipidemias
The hyperlipidemias (hyperlipoproteinemias) represent a group of metabolic diseases characterized by persistent elevation of plasma cholesterol levels, triglyceride levels, or both. Because plasma lipids circulate in the form of high molecular-weight complexes bound to protein, the term hyperlipidemia also indicates an elevation of lipoproteins, hence justification for the term hyperlipoproteinemia for this group of disorders. The dermatologic manifestation is the xanthoma, which may also be seen in metabolic disorders with normal levels of lipids (sitosterolemia and cerebrotendinous xanthomatosis) and disorders with deficiencies in high-density lipoproteins. Xanthomas can provide a clue that a child has a serious lipid abnormality and is at risk for other abnormalities, particularly vascular disease.
Lipid levels can be assayed in blood samples taken after a 12-hour fast. Plasma lipoproteins differ significantly in electrostatic charges, thus permitting their separation by electrophoretic mobility techniques into four major fractions: chylomicrons and β-, pre-β-, and α-lipoproteins. By means of ultracentrifugation it is also possible to separate the plasma lipoproteins into four major groups: chylomicrons and very low-density lipoproteins (VLDL; including the pre-β-mobility lipoproteins), low-density lipoproteins (LDL, the β-mobility lipoprotein), and high-density lipoprotein (HDL; the α-mobility lipoprotein), which correlate well with those separated by electrophoresis. Triglycerides are the major core lipids of chylomicrons and VLDLs. Cholesterol esters predominate in the core of LDLs, HDLs, remnants of VLDLs (also known as intermediate-density lipoproteins or IDLs ), and chylomicrons. Apolipoproteins mediate the binding of lipoproteins to their receptors in target organs and activate enzymes that metabolize lipoproteins ( Table 24-3 ). The levels of lipoproteins allow classification of the familial hyperlipidemias into five groups, designated as hyperlipoproteinemias I through V (World Health Organization [WHO]/ Frederickson classification system), each with its own specific clinicopathologic, prognostic, and therapeutic features ( Tables 24-4 and 24-5 ). Of these, types I and II most commonly present during childhood.
| Apolipoprotein | Association with Lipoprotein | Function |
|---|---|---|
| ApoA1 | HDL, chylomicrons | Main protein of HDL; activates lecithin/cholesterol acyltransferase |
| ApoA3 | VLDL, HDL, chylomicrons | Interacts with LDL receptorand affects lipoprotein metabolism |
| ApoB48 | Chylomicrons | Only in chylomicrons; ApoB100 without LDL receptor-binding domain |
| ApoB100 | LDL, VLDL | Main protein of LDL; binds to LDL receptor |
| ApoC2 | HDL, VLDL, chylomicrons | Activates lipoprotein lipase |
| ApoC3 | VLDL | Inhibits lipoprotein lipase and hepatic lisase to delay catabolism of triglyceride-rich particles |
| ApoE2, E3, E4 | HDL, VLDL, chylomicron remnants | Binds to LDL receptor |
| Disorder | Inheritance | OMIM No. | Prevalence | Cholesterol | Triglycerides | VLDL | Chylomicrons | LDL | HDL | Serum | Cause |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Type I | AR | 1/million | ↑ | ↑↑↑ | ↑ | ↑ | ↓ | ↓↓↓ | Creamy top | ||
| a: Familial hyperchylomicronemia | 239600, 246650, 615947 | a: Deficiency from mutations in lipoprotein lipase; LMF1 ; GPIHBP1 | |||||||||
| b: Familial apoprotein C2 or A-V deficiency | 207750, 133650 | b. Deficient ApoC2 or ApoA5 (see Type 5) | |||||||||
| c: — | 118830 | c. LP lipase inhibitor in blood | |||||||||
| Type II | 1 in 500 for heterozygotes | ↑ | NI or ↑ | ↑ | NI | ↓ | ↓ | Clear | |||
| a: Familial hypercholesterolemia | AD | 143890, 144010, 603776 | LDL receptor defect in 60%–80%; APOB, PCSK9, each <5% | ||||||||
| AR | 603813 | LDLRAP1 | |||||||||
| b: Familial combined hyperlipidemia | AD, AR | 144250 | 1 in 100 | Clear | Polygenic | ||||||
| Decreased LDL receptor and ApoB100 dysfunction | |||||||||||
| Type III | AR | 107741 | 1 in 10,000 | ↑ | ↑ | ↑ | ↑ | ↓ | NI | Turbid | ApoE2 synthesis |
| Familial dysbetalipoproteinemia | |||||||||||
| Type IV | AD | 144600 | 1 in 100 | ↑ | NI ↑ | ↑ | NI | ↓ | ↓↓ | Turbid | Renal disease, diabetes |
| Familial hypertriglyceridemia | |||||||||||
| Type V | AR | 144650 | Very rare | ↑ | ↑↑↑ | ↑ | ↑ | ↓ | ↓↓↓ | Creamy top, turbid bottom | Apo A-V (ApoA5) deficiency |
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


