Classification of Vascular Lesions
Vascular birthmarks, or congenital vascular anomalies, are common lesions that may present in a variety of fashions. There has traditionally been a significant amount of confusion regarding the nomenclature of these lesions, and the term hemangioma has been widely used in the medical literature in reference to a variety of different vascular anomalies. In 1982, Mulliken and Glowacki proposed a classification system for vascular birthmarks based on clinical and cellular features. This classification was further refined 6 years later in a book, and in 1996 it was adopted as the official classification system for vascular anomalies by the International Society for the Study of Vascular Anomalies (ISSVA). This nomenclature revolutionized the classification of vascular lesions, and is the basis for continued study into the causes of these lesions and their therapy.
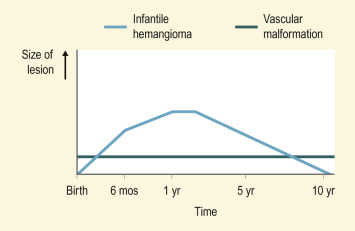
According to this classification system ( Box 12-1 ), vascular birthmarks are divided into the categories of tumors and malformations. Vascular tumors are neoplasms of the vasculature. This category includes hemangioma of infancy (the most common vascular tumor), kaposiform hemangioendothelioma (KHE), tufted angioma, and pyogenic granuloma (PG). Vascular malformations represent anomalous blood vessels without any endothelial proliferation or cellular turnover. In distinction to infantile hemangioma (IH), these lesions tend to be present immediately at birth and persist for a lifetime. Vascular malformations are further classified according to their predominant components, i.e., capillary malformation (port wine stain [PWS], salmon patch), venous malformation (VM), lymphatic malformation (LM), and arteriovenous malformation (AVM). Figure 12-1 pictorially demonstrates the different natural histories of IHs and vascular malformations.
Vascular tumors
Hemangioma of infancy
Tufted angioma
Kaposiform hemangioendothelioma
Pyogenic granuloma
Hemangiopericytoma
Vascular malformations
Capillary (CM) (i.e., port wine stain, salmon patch)
Venous (VM)
Lymphatic (LM)
Arterial (AM)
Arteriovenous (AVM)
Complex/combined (i.e., capillary–lymphatic–venous or capillary–venous)
This chapter includes discussion of vascular tumors and tumor syndromes, vascular malformations and malformation syndromes, disorders associated with vascular dilation, and a few miscellaneous disorders of the cutaneous vasculature.
Vascular Tumors and Tumor Syndromes
Infantile Hemangioma
IH, or hemangioma of infancy, is the most common benign soft-tissue tumor of childhood. Although estimates vary, it occurs in around 2% to 12% of infants. Female infants are three times more likely to have IHs than male infants, and the incidence is increased in premature neonates. Although they occur in all races, IHs appear to be less common in those of African or Asian descent. Multiple gestation pregnancy, advanced maternal age, placenta previa and preeclampsia also appear to be risk factors for IH. Although they are traditionally considered to be sporadic lesions, autosomal dominant segregation within families has been described. These lesions vary considerably in their appearance and significance, related to their size, depth, location, growth pattern, and stage of evolution. Older descriptive terms for IH include strawberry, cavernous , and capillary hemangiomas. These terms are no longer useful and should not be used in the era of more specific vascular lesion nomenclature, as discussed earlier and shown in Box 12-1 . Although IHs may occur on any part of the body, they most commonly involve the head and neck regions. Facial hemangiomas have been noted to have a nonrandom distribution, with the majority of lesions occurring on the central face at sites of development fusion. Four primary segments of facial IH distribution have been identified, as shown in Table 12-1 .
| Segment Number/Name | Distribution | Comment | |
|---|---|---|---|
| 1 | Frontotemporal segment | Lateral forehead | Potentially greater risk of associated brain anomalies when PHACES syndrome present |
| Anterior temporal scalp | |||
| Lateral frontal scalp | |||
| Upper eyelid | |||
| 2 | Maxillary segment | Lateral cheek | |
| Upper lip | |||
| Spares philtrum, preauricular area | |||
| 3 | Mandibular segment | Preauricular area | Potentially greater risk of cardiac defects when PHACES syndrome present |
| Mandible | |||
| Chin | |||
| Lower lip | |||
| 4 | Frontonasal segment | Medial frontal scalp/forehead | |
| Nasal bridge | |||
| Nasal tip/ala | |||
| Philtrum | |||
The pathogenesis of IH is not yet completely understood. Positive endothelial-cell staining with erythrocyte-type glucose transporter (GLUT)-1 protein has been noted in lesions during all of the growth phases. It is absent in other vascular lesions such as vascular malformations and hemangioendotheliomas. This protein is normally expressed in the microvascular endothelia of blood–tissue barriers such as the brain, retina, placenta, and endoneurium. As such it has been suggested that IH may originate from invading angioblasts that have differentiated toward a placental cell type or from embolized placental cells, although some data refute a placental trophoblastic origin for these lesions. Further evidence of a potential relationship between human placenta and IH includes the high level of transcriptome similarity between these tissues and the higher incidence of pathologic placental findings observed from pregnancies resulting in a child with IH compared with those resulting in healthy infants without IH. Other endothelial-cell markers shared between IH and placental tissue include Lewis Y antigen, FcγRII, and merosin. Recently, the Wilms tumor 1 ( WT1 ) gene was found to also stain proliferative vascular tumors, including IH. Aside from the placental embolization theory of pathogenesis, other hypothesized etiologies for IH development include a somatic mutation in a gene-mediating endothelial-cell proliferation or hypoxia-induced stimulation of endothelial progenitor cells.
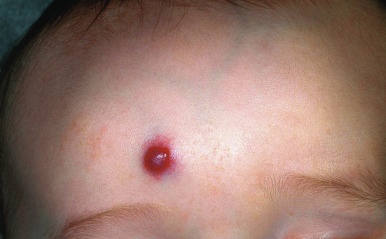
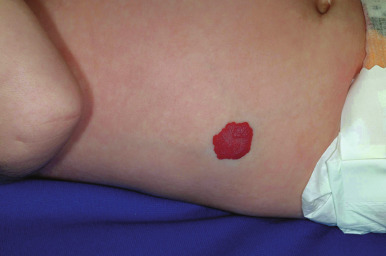
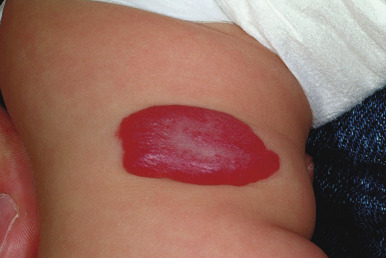
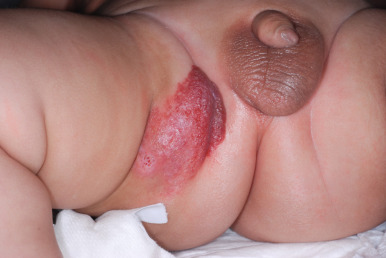
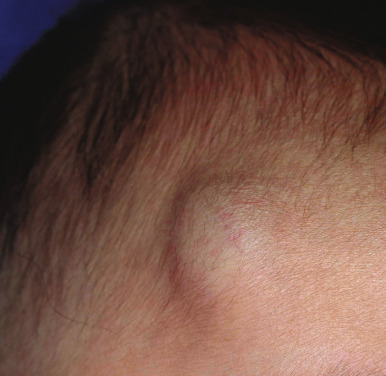
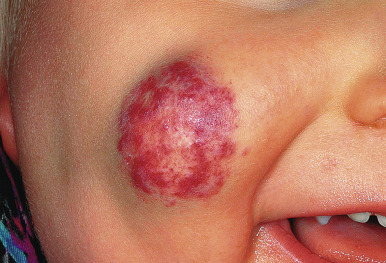
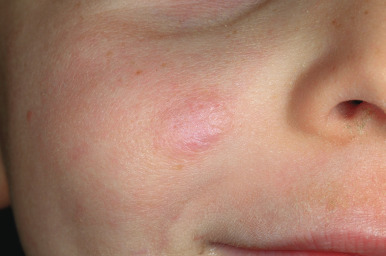
Hemangiomas may occur as superficial, deep, or mixed lesions representing their soft-tissue depth. The clinical appearance of the lesion depends on the type of hemangioma present. Superficial IHs, when well formed, present as bright red to scarlet, dome-shaped to plaque-like to lobulated papules, plaques, and nodules ( Figs. 12-2 through 12-6 ). They may partially blanch with pressure and are rubbery or noncompressible on palpation. Deep IHs usually present as subcutaneous, partially compressible nodules and tumors, often with an overlying blue hue, prominent venous network, or telangiectasias ( Figs. 12-7 through 12-9 ). These lesions may be warm to palpation. Combined IHs have both a superficial component and a deep component and occur in up to 25% to 30% of patients. They present with both the superficial, bright red component and a deeper, blue nodular component ( Figs. 12-10 and 12-11 ). Hemangiomas may also be divided into subtypes based on their anatomic configuration. Focal lesions are discrete, usually round lesions that appear to arise from a single focal point. Segmental lesions (see below) involve a more broad anatomic unit that may be determined by embryonic placodes. Indeterminate lesions are difficult to classify into one of the preceding two subtypes.
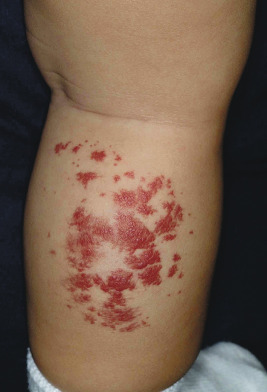
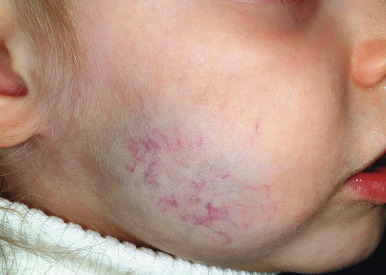
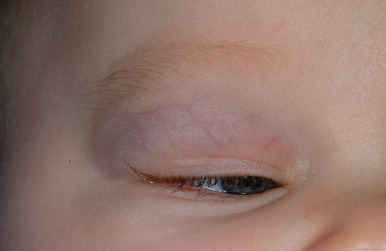
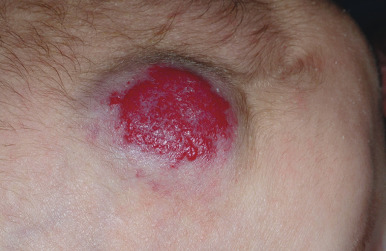
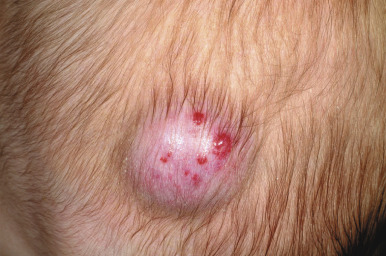
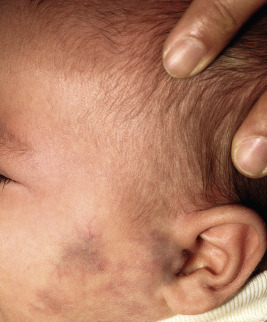
Hemangiomas may present with a variety of precursor lesions, and in up to 50% of affected infants these premonitory marks may be evident at birth. They include areas of telangiectasias ( Fig. 12-12 ), pallor, ecchymotic macules ( Fig. 12-13 ), and even ulceration ( Figs. 12-14 and 12-15 ). Fully formed IHs may occasionally present at birth and are termed congenital hemangiomas. These lesions are discussed in more detail later.
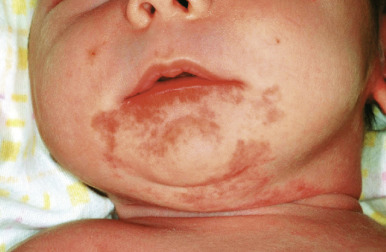
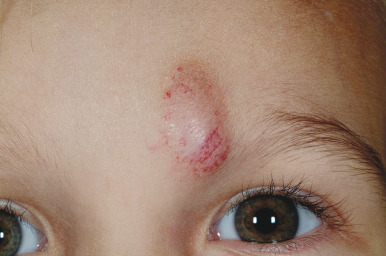
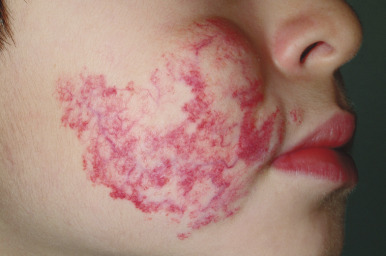
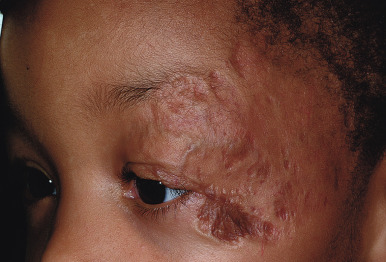
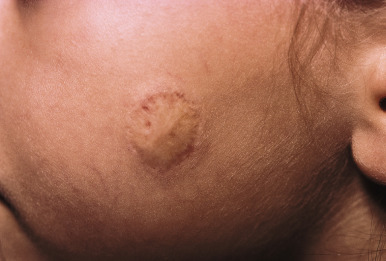
The natural history of IH is characteristic. It is notable for a period of growth (the proliferative phase), a period of stability (the plateau phase), and a period of spontaneous regression (the involutional phase). The majority of IHs first become evident at 2 to 3 weeks of life, with potential continued growth until around 9 to 12 months of age. The majority of growth, however, occurs during the first 5 months of age, and in those that continue to grow beyond this age, the growth rate is markedly slower. Occasional lesions have a proliferative phase that lasts for longer than 1 year, sometimes as long as 18 to 24 months. Deep IHs tend to exhibit both a delayed onset of growth as well as a sustained, longer growth phase. The onset of the involutional phase, which is difficult to predict in any given patient, is marked by a color change from bright red to dull red, purple, or gray (for superficial lesions) ( Figs. 12-16 and 12-17 ). It may be more difficult to appreciate early involution in deep IH, but with time these lesions become smaller, more compressible, and less warm. It is estimated that completed involution of IH occurs at a rate of 10% per year, such that 30% have involuted by 3 years of age; 50% by 5 years of age; 70% by 7 years of age, and more than 90% by 9 or 10 years of age. However, it should be remembered that involution does not necessarily imply totally normal skin. Possible residual changes after IH involution include telangiectasias ( Fig. 12-18 ), atrophy ( Fig. 12-19 ), scarring ( Fig. 12-20 ), or fibrofatty masses ( Figs. 12-21 and 12-22 ). These possibilities must be explained thoroughly to the parents of patients with IH.
Treatment decisions regarding an IH must incorporate many factors. These include the size and location of the lesion or lesions (see below), the age of the patient and growth phase of the hemangioma, associated findings, and the perceived potential for psychosocial distress both for parents and for the patient later in life. The latter is quite difficult to predict but must be seriously considered in the patient with a conspicuous IH, especially if it is facial. These lesions may be associated with parental disbelief, fear, mourning, and social stigmatization. A hemangioma severity scale has been developed and validated and incorporates the factors above, as well as risk for associated structural anomalies, complications, pain, and likelihood of disfigurement. The major goals of management should be to prevent or reverse life- or function-threatening complications; prevent disfigurement; minimize psychosocial stress; avoid overly aggressive procedures; and adequately prevent or treat ulceration in order to minimize infection, pain, and scarring. Treatment decisions can be challenging and must include parental input and a thorough risk-to-benefit analysis. Hemangiomas for which further evaluation, referral to a specialist, and/or therapy should be considered are listed in Box 12-2 . (These are discussed in more detail under Clinical Variants and Associated Syndromes.)
Life-threatening
Function threatening
Periocular
Nasal tip
Ear (extensive)
Lips
Genitalia, perineum
Airway
Hepatic
Large facial
Large anogenital/perineal
“Beard” distribution *
* See text for discussion.
Ulcerating
Lumbosacral
Multiple
Treatment options for IH are listed in Table 12-2 . Probably the most useful principle in the treatment of these lesions is that of active nonintervention. This term implies that the physician inquires about the parents’ knowledge of the condition, offers education regarding natural history and therapeutic indications, and offers anticipatory guidance and support. Active nonintervention is an appropriate “therapy” for the majority of IHs, and can be offered by the pediatrician or primary care physician in most instances. Serial photography may be useful for parents of children with hemangiomas in order to document the gradual spontaneous involution, which may be subtle and underappreciated by them. Referral to support groups and informational resources, such as the Hemangioma Investigator Group ( www.hemangiomaeducation.org ), Vascular Birthmarks Foundation ( www.birthmark.org ), or the National Organization of Vascular Anomalies ( www.novanews.org ), is useful for parents of these children.
| Treatment | Comment |
|---|---|
| Active nonintervention | Emotional support and guidance; distribution of patient education materials and referral to informational and support networks |
| Local wound care | When ulcerated: topical antibiotic, nonstick wound dressings, compresses, becaplermin gel |
| Oral antibiotics | When secondary infection present |
| Pain control | When ulcerated: may require temporary use of narcotic analgesics |
| Topical corticosteroids | Potent formulations; may be useful for localized, superficial IH |
| Intralesional corticosteroids | Usually triamcinolone; localized lesions; caution with periocular IH |
| Oral corticosteroids | Traditional gold standard for systemic therapy; prednisone or prednisolone, 2–4 mg/kg per day |
| Topical β-blockers | Most often timolol gel-forming ophthalmic solution, 0.1%–0.5%, applied 2–4 times daily; may be useful for smaller, localized and/or superficial IH |
| Oral β-blockers | Usually propranolol 2 mg/kg per day; risks include bradycardia, hypotension, hypoglycemia, bronchospasm, hypothermia, nightmares |
| Interferon α | Subcutaneous injection; severe, treatment-resistant, or life-threatening IH; risk of spastic diplegia |
| Laser therapy | Usually PDL; mainly useful for ulcerated IH or residual surface vascularity (i.e., telangiectasias) persisting after involution |
| Vincristine | Severe, treatment-resistant or life-threatening IH; requires venous access; risks include myelosuppression, peripheral neuropathy, extravasation necrosis |
| Surgical excision | Useful in select situations, i.e., incomplete resolution, disfiguring facial lesions, smaller complicated IH refractory to medical therapy |
Corticosteroids are the traditional mainstay of therapy for IHs requiring treatment. They are most often used in the oral form, although both intralesional and topical preparations may be useful. Intralesional corticosteroids are useful for localized lesions. Several injections may be necessary, and periocular lesions should be treated only by a physician experienced in their administration, given the possibility of embolization of corticosteroid particles and permanent vision loss. Topical corticosteroid therapy with clobetasol (a potent, class 1 topical steroid) may be useful for localized IH. In the authors’ experience, this therapy is most useful for macular or very thin plaque hemangiomas in the early proliferative stage. This medication should be used cautiously with attention to the risks of atrophy, ocular toxicity, and adrenal suppression.
Oral corticosteroids were at one time considered the gold standard for treatment of complicated IHs, and their mechanism of action is poorly understood. They are most useful during the proliferative phase and are generally administered in dosages ranging from 2 to 4 mg/kg per day of prednisolone or prednisone. Some authors advocate doses up to 5 mg/kg per day. This therapy is usually continued for several months, with gradual tapering as tolerated. If corticosteroids are tapered too quickly, rebound growth and adrenal suppression may occur. The goals of decreased IH growth or partial shrinkage must be balanced by the risks of long-term therapy. Common side effects include irritability, weight gain, hypertension, and gastrointestinal (GI) upset. Because the immune response is suppressed in patients receiving systemic corticosteroids, live-virus vaccinations should not be given during (or within 1 month after discontinuation of) therapy. Although decreased linear growth velocity may occur in treated patients, catch-up growth usually occurs after their discontinuation. Adrenal axis suppression is rare, and when it occurs, tends to be reversible. Many experts treat concomitantly with H2 blockers (i.e., ranitidine) to diminish the potential for symptomatic steroid-associated gastritis or worsening of gastroesophageal reflux. Pneumocystis carinii pneumonia has been rarely reported, which has prompted some experts to recommend trimethoprim-sulfamethoxazole prophylaxis during systemic steroid therapy. In the more recent era of β-blocker therapy for IH, oral corticosteroids are used significantly less often.
Interferon α-2a or -2b, a potent inhibitor of angiogenesis, has been used successfully in patients with IH refractory to corticosteroid therapy. This medication is administered as a daily subcutaneous injection, in a dose of 1 to 3 million units/m 2 . Common side effects include fever, irritability, and malaise. Neutropenia, anemia, and elevation of hepatic transaminases may also occur. However, the most concerning toxicity of this therapy is neurologic in origin, most notably spastic diplegia, which may occur in up to 20% of treated patients. Because of this risk, interferon therapy should be reserved for patients with severe life- or function-threatening lesions that are recalcitrant to other therapies, and neurologic evaluation before and during therapy should be performed. The chemotherapeutic agent vincristine is also occasionally used for steroid-resistant, life-threatening IHs.
The current standard of care for IH requiring systemic therapy is propranolol. This nonselective β-blocker, which is traditionally used for cardiac indications such as hypertension or arrhythmia, was incidentally noted to result in marked shrinkage of IHs when administered in two children who had a cardiac indication and who also happened to have hemangiomas. Subsequently these effects were confirmed in several more patients reported by the same authors as well as in several other open-label series, prospective studies, and retrospective reviews In the years after the initial report, propranolol has evolved to become the therapy of choice for most clinicians with expertise in the treatment of IH. A multicenter, prospective, international, placebo-controlled study of oral propranolol for IH has been completed and culminated in the recent approval of propranolol for treating IH by the US Food and Drug Administration as well as the European Medicines Agency. The mechanism of action of propranolol in this setting is unclear, although most attribute the effects to some combination of vasoconstriction, inhibition of proangiogenic growth factors (including vascular endothelial growth factor, basic fibroblast growth factor, and matrix metalloproteinase-9), and induction of apoptosis. It is most often started at around 0.5 to 1 mg/kg per day, with the dosage gradually titrated up to 2 to 3 mg/kg per day divided into two to three doses daily. Potential side effects include hypotension, bradycardia, bronchospasm, hypoglycemia, hypothermia, and nightmares, and this agent should be administered under close supervision with attention to these possible toxicities. Rebound growth or recurrence of IH after propranolol therapy has been increasingly recognized and may occur in 6% to 19% of patients. The time between cessation of therapy and notation of rebound is typically 3 to 6 months, and most lesions respond well to a second course of propranolol. Although propranolol therapy tends to be most effective during the early proliferative phase of IH (i.e., first 6 months), lesions in the postproliferative phase may also respond to treatment.
Hypoglycemia has surfaced as one of the most relevant (and preventable) potential toxicities of oral propranolol therapy, and the highest risk period for this effect is in the morning after the overnight fast that occurs during sleep. The mechanism for the development of hypoglycemia is likely related to β-blocker-mediated inhibition of gluconeogenesis and glycogenolysis, mandatory components of physiologic glucose homeostasis. Measures to decrease this risk include always giving propranolol with food, holding therapy during periods of illness or decreased oral intake, and the avoidance of fasting for longer than 8 hours (which may require nighttime awakenings for feeding). Parents should be taught to recognize early warning signs of hypoglycemia, including shakiness, sweating, anxious appearance, and coldness. Table 12-3 list some guidelines for the use of propranolol in the treatment of IH.
| Guideline | Comment |
|---|---|
| Consider contraindications | Cardiogenic shock, sinus bradycardia, hypotension, heart block (>1st degree), heart failure, bronchial asthma, hypersensitivity to propranolol |
| Perform ECG for patients at risk | At-risk category includes: HR below normal for age, family history of congenital heart conditions or arrhythmias, maternal history of connective tissue disease, personal history of arrhythmia or one noted during examination |
| Patients with PHACES syndrome: thorough imaging evaluation of head, neck, and cardiac arterial anatomy should be performed before initiation of therapy | If imaging findings place patient into higher risk for stroke, consult/comanage with neurologist during therapy; use lowest effective dose possible |
| Inpatient initiation of therapy recommended for infants ≤8 weeks’ gestationally corrected age, with comorbid conditions, or with poor social support | Outpatient initiation of therapy appropriate for infants >8 weeks’ gestationally corrected age, with adequate social support, and lack of comorbid conditions |
| Typical dosing is 1 mg/kg per day (initiation) and 2–3 mg/kg per day (maintenance), divided three times daily | BP and HR checks recommended at baseline, 1 and 2 hours after initial dose, and repeated with each dose increase of >0.5 mg/kg per day (until maintenance dose reached); dose may be increased every 3–7 days until goal dosage is reached |
| Always dose propranolol with food and avoid prolonged fasting | Discontinue during illness or periods of decreased oral intake; awaken baby to feed at night as needed to prevent sleeping longer than 8 hours without eating |
| Hold propranolol during illness that includes wheezing | β-blockers may worsen bronchospasm |
After the discovery of propranolol’s efficacy in treating IH, several investigators reported the use of a topical nonselective β-blocker timolol for small, uncomplicated superficial hemangiomas. Topical timolol maleate gel-forming ophthalmic solution is classically used for the treatment of glaucoma, but increasing reports have described its efficacy in subsets of patients with these smaller IH. Most often used in concentrations of 0.1%, 0.25%, or 0.5%, it is applied two to four times daily to the surface of the IH and rubbed in well. Most small, superficial IHs and even some combined (superficial and deep) lesions respond to the treatment, which in general is very well tolerated. Systemic β-blocker side effects have been noted to occur only rarely, although adequate parent education about these potential toxicities (see previous discussion of propranolol) is warranted. Topical timolol ophthalmic solution is a reasonable consideration for small, primarily superficial, uncomplicated, and functionally insignificant IH.
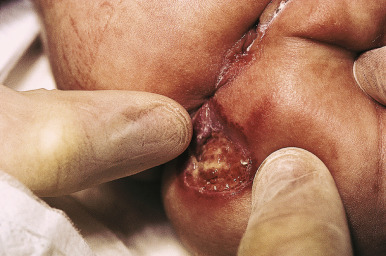
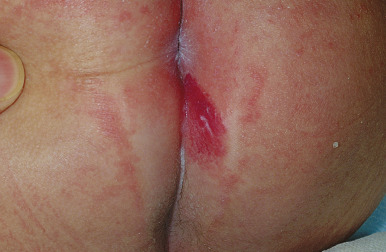
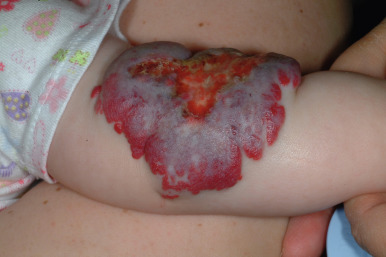
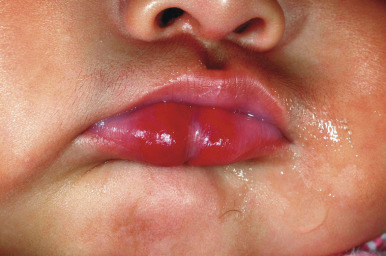
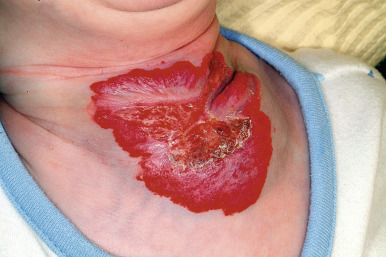
Ulcerated IH deserves special mention, as this is the most common complication in these lesions, occurring in up to 16% of patients. Ulceration usually occurs during the proliferative phase ( Fig. 12-23 ) and is especially common in areas of recurrent trauma such as the lips ( Fig. 12-24 ), genitals ( Fig. 12-25 ), perineum/perianal region, or in moist intertriginous sites such as the neck ( Fig. 12-26 ). Tissue breakdown results in bleeding, secondary infection, pain, and permanent scarring. Local wound care with compresses, topical antibiotics (i.e., bacitracin, mupirocin, or metronidazole), and nonstick wound dressings (i.e., petrolatum-impregnated gauze) are useful. Oral antibiotics should be considered if persistent or deep ulceration, drainage, or exudate is present and should be guided by the results of culture and sensitivity testing. Pain management with acetaminophen or acetaminophen with codeine may be indicated. Topical lidocaine may be helpful for pain control but should be used with extreme caution given the risks of absorption and possible systemic toxicity. Becaplermin gel, a recombinant human platelet-derived growth factor approved for topical therapy of lower-extremity diabetic neuropathic ulcers in patients 16 years of age and older, has been used off-label for the treatment of ulcerated IH with good success. Ulcerated IHs that do not improve with the above treatments usually warrant systemic therapy.
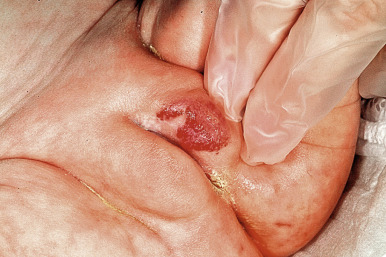
Pulsed-dye laser (PDL; the word laser is an acronym for light amplification by stimulated emission of radiation ) therapy may be useful in the treatment of ulcerated IH. This laser emits a wavelength of light specific for oxyhemoglobin, thus imparting specificity for vascular structures (selective photothermolysis). Because the light penetrates only 1 mm of depth in the skin, this modality is generally not helpful for nonulcerated, elevated, or nodular lesions. With ulcerated lesions, though, it is useful in accentuating reepithelialization and decreasing pain. The choice of PDL therapy must be individualized to the patient, and occasionally patients may show no response or even worsening with this modality. PDL therapy may also be useful for residual surface vascularity after IH involution. Although PDL is the most commonly utilized form of laser therapy for IH, other lasers such as the neodymium-doped yttrium aluminum garnet (Nd:YAG) laser or the alexandrite laser may offer some benefit but carry a higher risk of scarring.
Clinical Variants and Associated Syndromes
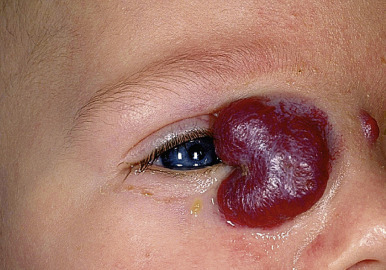
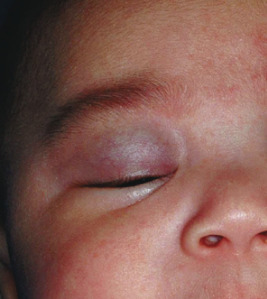
Hemangiomas in certain locations may suggest a higher probability of complications or associated findings. Hemangiomas involving various portions of the face may be quite problematic. Periorbital IHs may be relatively insignificant when small, but larger lesions ( Fig. 12-27 ) may result in a variety of ocular complications. In addition, any periorbital IH may be indicative of deeper, retrobulbar involvement, which may or may not present with unilateral proptosis. Light-deprivation amblyopia may result from visual axis obstruction, most commonly in the setting of significant upper eyelid lesions that result in mechanical ptosis ( Fig. 12-28 ). Astigmatism may occur if the IH compresses the globe and may result in permanent amblyopia. Rarely, IH situated near a tear duct may block the drainage system and result in matting and conjunctivitis. Because of these concerns, any child with a periorbital IH should undergo ophthalmologic examination. If there are functional or anatomic ophthalmologic concerns, systemic therapy is usually indicated.
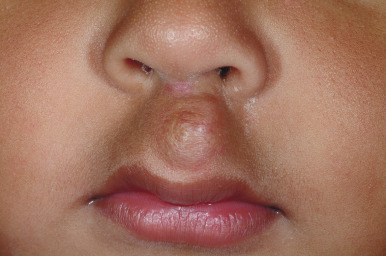
Nasal tip/bridge IHs ( Fig. 12-29 ) often require specialist consultation because of their propensity to distort the nasal anatomy ( Fig. 12-30 ), resulting in permanent disfigurement. Slow involution of these lesions may pose significant psychosocial distress to the older child, and the splaying of nasal cartilage combined with redundant fibrofatty tissue after involution may result in the “Cyrano” nasal deformity ( Fig. 12-31 ). The nasal crease sign is a linear, gray atrophic crease in the inferior columella that may occur with segmental facial IH involving the nose ( Fig. 12-32 ) and that may portend imminent cartilage destruction and nasal collapse. It is for reasons such as these that therapy is often indicated for nasal IH.
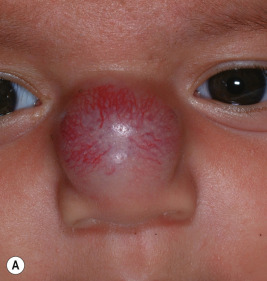
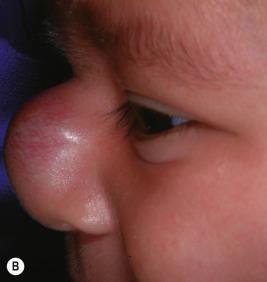
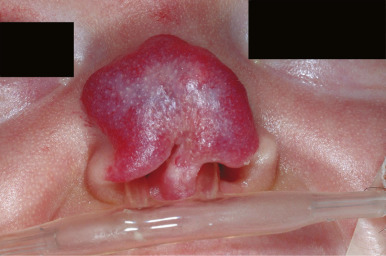
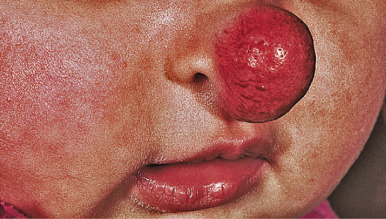
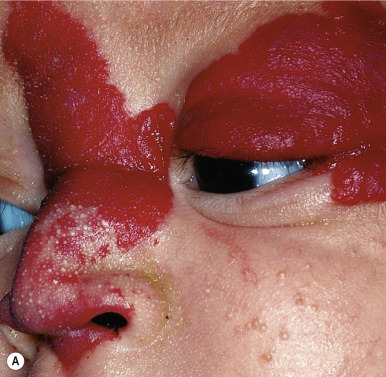
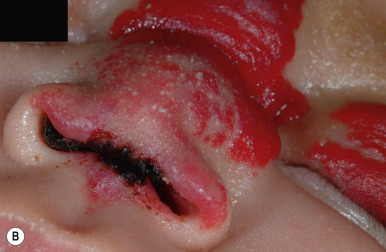
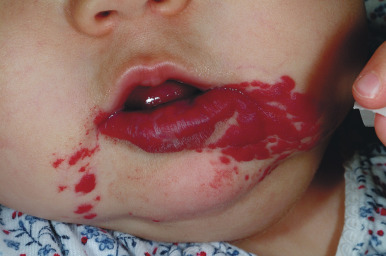
Hemangiomas located on the lips tend to involute slowly and are more prone to ulceration and bleeding. These changes may in turn contribute to increased pain, feeding difficulties, and permanent scarring. Ear IHs may also be cosmetically disfiguring, and when large may obstruct the auditory canal and result in conductive hearing loss.
Hemangiomas that involve the neck, lower lip, chin, preauricular, and mandibular areas (beard lesions) ( Fig. 12-33 ) are clinically important because of the increased risk of associated airway lesions in these patients. In one study, patients with more extensive IHs in this location had up to a 63% incidence of subglottic or upper-airway hemangiomatosis. Airway IH may also occur in the absence of cutaneous IH. Infants with symptoms are usually brought for treatment within the first few months of life with croup-like cough, hoarseness, and biphasic stridor. Any infant with extensive IH in this distribution and/or symptoms of upper-airway obstruction should be immediately referred for direct visualization of the airway with laryngoscopy. Treatment options in this setting may include tracheotomy, propranolol, corticosteroids, interferon α, surgical excision, and laser therapy.
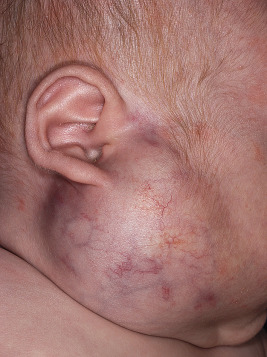
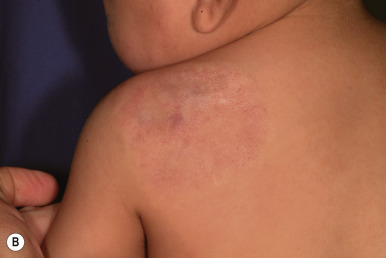
Parotid IHs often present as mixed superficial and deep lesions with a prominent deep component of swelling with an overlying blue hue ( Fig. 12-34 ). These lesions tend to grow significantly and may involute more slowly than IHs in other locations. They may have a poorer response to pharmacologic therapy, although propranolol has been demonstrated to be effective in many patients. Genital ( Fig. 12-35 ) and perineal IHs are problematic because of ulceration, with the concomitant risks of bleeding, infection, permanent scarring, and pain. When these lesions are larger and/or segmental, the possibility of associated developmental anomalies of the external genitalia, anorectal region, genitourinary tract, spine and spinal cord must be considered (see PELVIS Syndrome). Hepatic IHs may be associated with significant morbidity and mortality and are discussed further in the Diffuse Neonatal Hemangiomatosis section.

Lumbosacral IHs ( Fig. 12-36 ) are important, because they may signal underlying occult spinal dysraphism or spinal cord defects. Tethering of the spinal cord is one of the more common associations, and this anomaly may result in permanent neurologic sequelae without release. Other common findings include spinal lipoma, intraspinal hemangioma, and sinus tracts. In a prospective study of 41 patients with lumbosacral IHs who underwent magnetic resonance imaging (MRI) examination, intraspinal abnormalities were detected in 21 patients, with a positive predictive value of IH for spinal dysraphism of 51.2%. An increased risk for spinal anomalies correlated with IH ulceration and the presence of additional cutaneous anomalies (including gluteal cleft asymmetry or deviation, sacral dimple, skin appendage, lipoma, dermal melanocytosis, dermal sinus, and aplasia cutis). Importantly, 35% of patients with an isolated lumbosacral IH had spinal dysraphism. Also important, 42% of children with normal ultrasound scans who then underwent MRI evaluation had spinal anomalies, highlighting the potential risk of relying on ultrasonography as a screening tool for high-risk infants. In addition to spinal anomalies, sacral IH may also be associated with various congenital anorectal or urogenital anomalies, as discussed with genital and perineal lesions. This association is discussed in more detail below (see PELVIS syndrome). Any infant with a lumbosacral IH, especially when it is large or of the segmental type or when other cutaneous stigmata are present, should be considered for MRI to assess for underlying abnormalities.
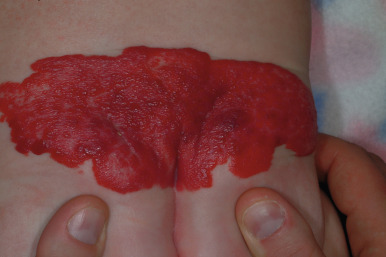
Segmental hemangiomas are lesions that involve a broad anatomic region (see Fig. 12-36 ; Fig. 12-37 ) or a recognized developmental unit. They are often unilateral and sharply demarcated at the midline, although exceptions occur. Segmental IHs are most commonly located on the face. They seem to have a higher rate of complications and entail a greater risk for functional compromise and associated abnormalities such as urogenital anomalies or the posterior fossa malformations, hemangioma, arterial anomalies, cardiac defects, eye anomalies, and sternal defects (PHACES) syndrome (see the following paragraph). In addition, they are more often complicated by ulceration. The association of segmental IH with GI (typically lower GI) bleeding has been highlighted and appears to occur most often in the setting of PHACES syndrome with aortic dysplasia.
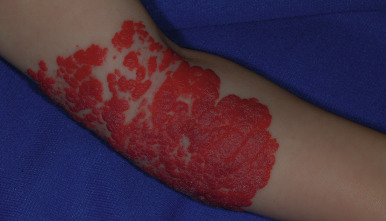
The PHACES syndrome or PHACES association (Online Mendelian Inheritance in Man, 606519) is a constellation of clinical findings associated with extensive facial IH. Table 12-4 outlines the various features seen in this syndrome. The majority of patients with PHACES syndrome are female. Although the pathogenesis of this syndrome is unclear, it appears to be a developmental “field defect” that arises around the seventh to tenth week of gestation. The facial IHs in patients with PHACES syndrome are usually large and segmental ( Fig. 12-38 ) and may be unilateral or bilateral. They usually follow an aggressive growth pattern, and ulceration is common. There may be a dermatomal distribution, although these dynamic lesions should not be confused with the static vascular malformations (PWSs) associated with Sturge–Weber syndrome (see below). When there is “beard” area involvement, airway hemangiomatosis should be considered. Hemangiomas may also occur in other nonfacial locations in the setting of PHACES syndrome, most notably the upper torso and extremities. Patients with large facial IHs, especially in the presence of ventral developmental defects such as sternal clefting or abdominal raphe, should be screened for this syndrome with MRI of the brain, echocardiography, ophthalmologic examination, and close neurologic and head circumference examinations. Direct airway visualization should be considered if the infant has extensive involvement in the beard area or symptoms of upper airway obstruction.
| Manifestation(s) | Comment | |
|---|---|---|
| P | Posterior fossa malformations | Dandy–Walker malformation; cerebellar atrophy; hypoplasia or agenesis of various CNS structures; may also have intracranial hemangioma, midline anomalies, neuronal migration disorder |
| H | Hemangioma | Extensive facial; plaque-like; segmental; occasional airway involvement |
| A | Arterial anomalies | Mainly head and neck; aneurysms, anomalous branches, aberrancy, hypoplasia, absence, stenosis, kinking, looping, tortuosity; increased risk of AIS |
| C | Cardiac anomalies and aortic coarctation | PDA, VSD, ASD, PS, TF, others; in addition to aortic coarctation, may have dysplasia, aneurysm, aberrant origin of the subclavian artery with or without vascular ring |
| E | Eye abnormalities | Microphthalmia, optic-nerve dysplasia, persistent fetal vasculature, morning-glory disc anomaly; also Horner syndrome, retinal vascular abnormality, cataract, coloboma |
| S | Sternal clefting and supraumbilical abdominal raphe | Ventral midline developmental defects; may also include hypopituitarism, ectopic thyroid (with risk of hypothyroidism) |
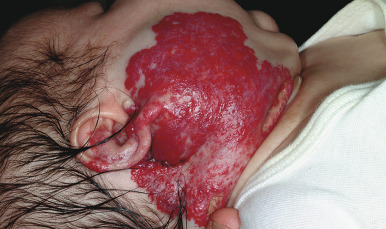
Anomalies of the cervical and cerebral vasculature are commonly noted in PHACES syndrome, and these patients appear to have a progressive arterial vasculopathy with risk of arterial ischemic stroke (AIS). Regressive changes in the vascular anomalies have also been noted in some patients. Common central nervous system (CNS) structural abnormalities include Dandy–Walker malformation, cerebellar hemisphere hypoplasia (ipsilateral to the hemangioma), and arachnoid cysts. Aortic arch anomalies, especially aortic coarctation, are the most commonly noted cardiac abnormalities in PHACES syndrome and tend to be complex and anatomically atypical. These anomalies may remain asymptomatic in the first days of life until the ductus arteriosus closes, at which time they may result in severe hemodynamic consequences. Box 12-3 lists the recommended evaluation for the infant deemed to be at risk for PHACES syndrome. Diagnostic criteria for PHACES syndrome have been published.
Baseline
Thorough physical examination, including:
Cutaneous (including assessment for ventral developmental defects)
Cardiac (including pressure measurements in all four extremities)
Respiratory (i.e., for stridor, hoarseness of cry, tachypnea)
Neurologic (i.e., for hypotonia, developmental delay)
Abdominal (i.e., for hepatomegaly, abdominal bruit)
MRI/MRA of the brain and neck
Echocardiography
Ophthalmologic evaluation
Thyroid function testing
If Indicated
MRI/MRA of the chest
Other endocrine evaluations (i.e., for growth hormone deficiency, hypopituitarism, diabetes insipidus)
Airway evaluation/direct laryngoscopy
MRI, Magnetic resonance imaging; MRA, magnetic resonance angiography; PHACES, posterior fossa malformations, hemangioma, arterial anomalies, cardiac defects, eye anomalies, and sternal defects.
PELVIS syndrome has been used to describe infants with perineal IH in association with a variety of other malformations involving the genitourinary and GI tracts, lumbosacral spine, and spinal cord. The acronym PELVIS stands for p erineal hemangioma, e xternal genitalia malformations, l ipomyelomeningocele, v esicorenal abnormalities, i mperforate anus, and s kin tag. This association has also been known as SACRAL syndrome , standing for s pinal dysraphism, a nogenital anomalies, c utaneous anomalies, and r enal and urologic anomalies associated with a ngioma of l umbosacral localization. More recently this phenotype has been expanded to include IHs involving not only the perineum but also the lumbosacral region, genitalia, and/or lower extremity by a group that suggested the terminology of LUMBAR association , for l ower body IH and other cutaneous defects, u rogenital anomalies/ u lceration, m yelopathy, b ony deformities, a norectal malformations/ a rterial anomalies, and r enal anomalies. In this series, the IHs were noted to often be segmental as well as of the “minimal growth” phenotype ( Fig. 12-39 ; also see below). Other cutaneous findings that may be present include lipoma, skin tag, caudal appendix, hair tuft, and sacral dimple. PELVIS/SACRAL/LUMBAR syndrome has been likened to PHACES syndrome, although it affects the lower half of the body and should be considered in any infant with a segmental IH (including those with minimal growth) affecting the anogenital, lumbosacral, or lower-extremity regions.
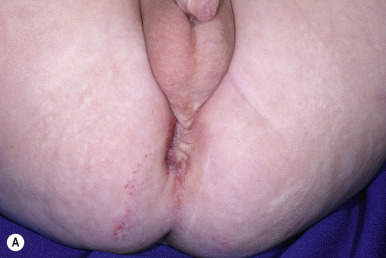
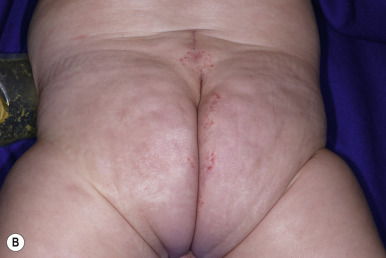
Abortive hemangiomas refers to IHs that exhibit very little (if any) proliferation during the period of expected growth. These lesions typically present as reticulated vascular patches, often with fine telangiectasias, occasionally as grouped telangiectases overlying normal-appearing skin, or as blue patches with a peripheral halo. Small foci of typical bright red, superficial IH may be present around the periphery ( Fig. 12-40 ). Other terms that have been used to describe these lesions include plaque-telangiectatic hemangiomas, macular hemangioma with PWS-like appearance, reticular IH, minimal-growth hemangiomas, and IHs with minimal or arrested growth ( IH-MAGs ). Several authors have confirmed these lesions to be IH in origin via positive GLUT-1 immunostaining of biopsies. IH-MAGs tend to present more commonly than typical IHs on the lower half of the body and may be associated with ulceration, especially when occurring in the anogenital area.
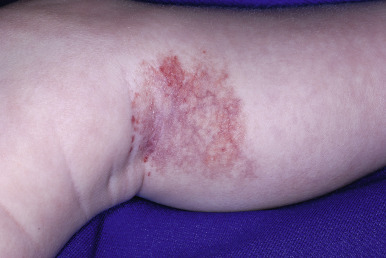
Congenital IHs are lesions that are fully formed at birth. These lesions may follow the typical time course of IH, may have a more accelerated involution phase, or may persist unchanged. Noninvoluting congenital hemangioma (NICH) is a specific subtype of IH also known as a congenital nonprogressive hemangioma. These lesions may be diagnosed in utero , are fully formed at birth, and do not show the postnatal proliferation characteristic of IH. Clinically NICH lesions are round to ovoid, pink to purple tumors with overlying prominent telangiectasias and peripheral pallor ( Fig. 12-41 ). Importantly, these lesions also do not undergo the spontaneous involution typical of IH and hence appear to be a distinctive type of vascular lesion. Interestingly, NICH lesions also have a different histologic appearance and are not immunoreactive for GLUT-1.
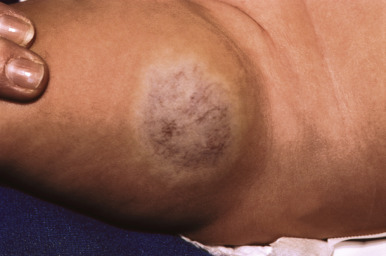
Rapidly involuting congenital hemangioma ( RICH ) is the term used to describe a congenital IH that undergoes rapid involution early in life. These tumors may present similar to an NICH or they may have the appearance of a typical IH. They may also present as small to large tumors with a red, violaceous, or gray appearance that may mimic other vascular (i.e., KHE, see Kaposiform Hemangioendothelioma section) or malignant (i.e., rhabdomyosarcoma, fibrosarcoma) neoplasms. They often involute completely by 12 to 15 months of age ( Fig. 12-42 ) and may leave behind dermal and subcutaneous atrophy.
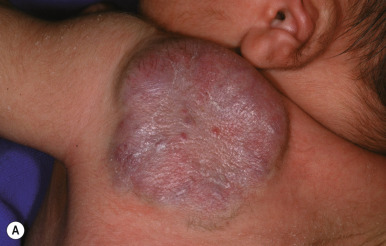
Diffuse Neonatal Hemangiomatosis
Diffuse neonatal hemangiomatosis (disseminated neonatal hemangiomatosis) describes patients with multiple cutaneous lesions in conjunction with extracutaneous organ involvement. It must be differentiated from benign neonatal hemangiomatosis, in which the infant has multiple cutaneous lesions without any symptomatic visceral lesions or complications. Patients with either type of hemangiomatosis may have only a few or up to hundreds of cutaneous IHs ( Figs. 12-43 and 12-44 ). Some authors advocate for the terminology multifocal IH with or without extracutaneous disease, noting that many cases reported in the literature as diffuse neonatal hemangiomatosis may actually represent other entities such as multifocal lymphangioendotheliomatosis with thrombocytopenia (MLT) (see later this chapter).
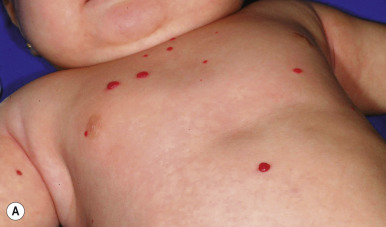
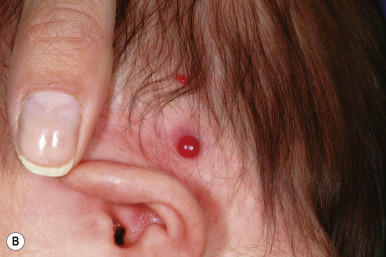
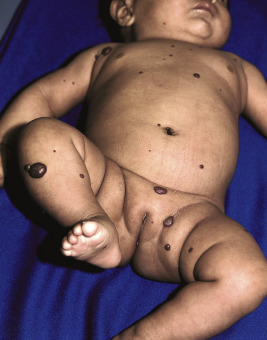
Although past data suggested that the risk of internal involvement could be predicted by the number of cutaneous IHs, the traditional recommendation for internal imaging in patients with five or more lesions was more anecdotal than evidence-based. Recently, however, in a prospective study comparing 151 infants with five or more IHs to 50 infants with between one and four IHs, hepatic hemangiomas were identified on hepatic ultrasound in 16% (24) of the infants with five or more cutaneous IHs and in none of the control group with one to four cutaneous IHs. The study also revealed that preterm (<37 weeks’ gestational age) delivery, multiple gestation, lower birth weight, in vitro fertilization, placental anomalies, and preeclampsia each occurred more often in infants with more than five cutaneous IHs, but on multivariate analysis only preterm delivery and lower birth weight were associated with having five or more cutaneous IHs. Of the 24 infants with hepatic hemangiomas, only two required therapy specifically for this finding. This study helps to confirm that infants with five or more IHs are at greater risk for hepatic hemangiomas (and should be evaluated with liver ultrasound) and that the majority of those with hepatic involvement remain asymptomatic and do not require therapy.
Although the most common extracutaneous site for IH in patients with diffuse hemangiomatosis is the liver, multiple other organs can be involved, including the intestine, brain, eyes, spleen, oral mucosa, kidney, and lungs. The diagnosis of hepatic hemangiomatosis is usually made clinically and radiographically, because liver biopsy is undesirable owing to the risk of bleeding. Liver lesions may be asymptomatic or present with hepatomegaly and congestive heart failure, the latter of which is believed to be the result of arteriovenous (AV) shunting, which leads to increased venous return and increased cardiac output. Occasionally anemia or thrombocytopenia is present.
Three patterns of liver hemangiomatosis have been proposed: focal, multifocal, and diffuse. Focal lesions are well defined and solitary on imaging and are most often clinically asymptomatic. They usually are not accompanied by cutaneous lesions and tend to involute rapidly. Multifocal liver hemangiomas present as multiple spherical tumors, which may be asymptomatic or can result in high-output cardiac failure secondary to AV shunting. These lesions involute after the same course as seen with cutaneous IH. Diffuse lesions are extensive, may result in near-total replacement of hepatic parenchyma, and may be associated with serious complications including vessel compression, respiratory decompensation, abdominal compartment syndrome, and multiorgan system failure. These lesions may also be associated with severe hypothyroidism.
Abdominal ultrasonography with Doppler studies is the diagnostic modality of choice, although MRI or computed tomography (CT) may occasionally be necessary. Treatment options may include optimization of cardiac function, corticosteroids, propranolol, interferon α, vincristine, cyclophosphamide, embolization, hepatic artery ligation, hepatic resection, and liver transplantation. Propranolol has been increasingly reported as an effective therapy for hepatic IH, and in most patients improvement in the liver lesions correlates with resolution of heart failure and improvement or resolution in the associated hypothyroidism, when present. Infants with diffuse liver hemangiomas should undergo thyroid hormone monitoring and replacement as necessary. Most visceral hemangiomas, like the cutaneous lesions, grow during infancy and in surviving children regress during early childhood. Other evaluations that may be useful in the investigation for diffuse hemangiomatosis include complete blood cell counts, stool examinations for occult blood, liver function studies, urinalysis, echocardiography, electrocardiography, and CNS imaging when clinically indicated.
Kasabach–Merritt Phenomenon
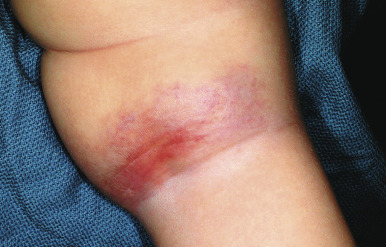
Kasabach–Merritt phenomenon (KMP; also known as Kasabach–Merritt syndrome ) is characterized by thrombocytopenia (owing to platelet trapping), coagulopathy, and microangiopathic hemolytic anemia in association with a rapidly enlarging vascular lesion. It was traditionally felt to be a complication of IH but is now known to be associated with vascular tumors other than IH, namely KHE and tufted angioma. These lesions have characteristics that clinically distinguish them from hemangiomas (see below). KMP occurs most often within the first few weeks of life and presents with sudden enlargement of the vascular lesion, ecchymosis, prolonged bleeding, epistaxis, hematuria, or hematochezia. The cutaneous lesions show purpura, edema, induration, and an advancing ecchymotic margin ( Fig. 12-45 ). In addition to thrombocytopenia, other laboratory findings include anemia, hypofibrinogenemia, elevated D-dimers, fragmentation of erythrocytes on manual smear, and prolonged coagulation studies. The mortality rate is high, from 10% to 30%.
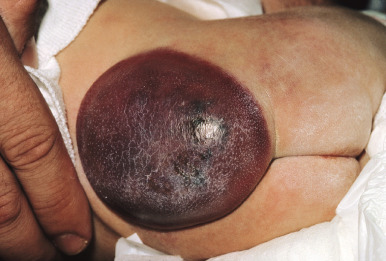
Kaposiform Hemangioendothelioma
Kaposiform hemangioendothelioma (KHE) is a locally aggressive vascular tumor, so named because of the histologic resemblance to Kaposi sarcoma. It presents as a firm, subcutaneous nodule or plaque that expands rapidly and often has a violaceous discoloration ( Fig. 12-46 ). Although KHE usually presents in infancy, it may appear months to years later. In addition to involvement of the skin, KHE may involve the deep soft tissues and the bone.
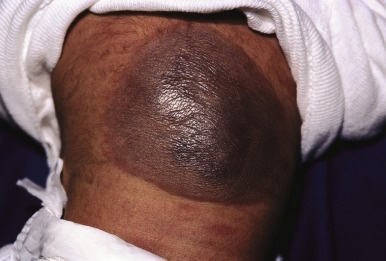
Tufted Angioma
Tufted angioma is actually named after its histologic appearance, which reveals groups of dermal capillary tufts. These lesions present clinically as erythematous, annular nodules, or plaques, often with induration ( Fig. 12-47 ). Although these lesions may spontaneously involute over years, they tend to persist or gradually progress in most patients. Both KHE and tufted angioma are usually easily distinguished clinically from IH, although in some patients either of the former two may mimic the latter.
Treatment for KMP is challenging. Multiple different regimens have been reported without randomized studies to support or refute their efficacy. It seems clear that a multidisciplinary approach is optimal, including medical and surgical components of diagnosis and therapy. Hematologic therapies include red blood cell transfusions and administration of fresh frozen plasma and cryoprecipitate. Many experts recommend minimization of platelet transfusions given the risks of further platelet trapping and further expansion of the tumor. High-dose corticosteroids, antifibrinolytics, interferon α, vincristine, sirolimus, cyclophosphamide and antithrombotics have been advocated. Vincristine was demonstrated useful in a retrospective study of 15 patients with KMP based on increased platelet count and fibrinogen level and decreased size of the tumor. Four of these patients had a relapse and were again treated successfully with the agent. Vincristine in combination with corticosteroids is considered first-line treatment for many experts. Combination vincristine and ticlopidine, without or with aspirin (vincristine-aspirin-ticlopidine [VAT] therapy), has been reported useful in several patients with KMP. Early surgical intervention with full resection has been advocated for smaller lesions where this modality is feasible. Other reported treatments include embolization, compression, and radiation therapy. Propranolol was associated with clinical response in only 25% of patients with KMP in one series, suggesting the need for further study of this agent.
Pyogenic Granuloma
Pyogenic granuloma (PG) is a common acquired vascular lesion of the skin and mucous membranes in infants and children. It presents as a bright red to red-brown, raised, slightly pedunculated or sessile papulonodule ( Figs. 12-48 and 12-49 ). The base of sessile lesions may reveal a peripheral collarette of scale ( Fig. 12-50 ). Although its appearance is usually solitary, PG may occasionally present as several lesions. In addition, secondary PG-like proliferations may occasionally occur on the surface of an existing PWS ( Fig. 12-51 ), usually in the older child or young adult. Multiple, or “agminated,” PG lesions have been reported in the setting of preexisting congenital vascular stains as well as in areas of preceding scalding burns. PG is prone to superficial ulceration and bleeding, which usually brings the patient to medical attention. The pathogenesis of PG is unknown. Despite the name, the lesions are not considered infectious. Although they may occur on any skin surface, they most commonly appear in areas subject to trauma, especially the hands, fingers, forearms, face, and occasionally the mucosal surfaces of the mouth. They have been described on the penile shaft after circumcision. Although histologic evaluation is usually not necessary for diagnosis, pathologic examination reveals changes similar to those of a well-circumscribed IH.
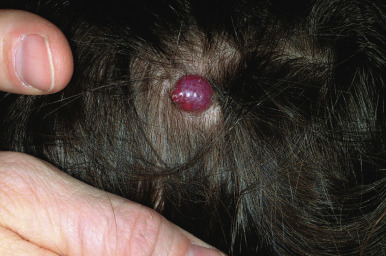
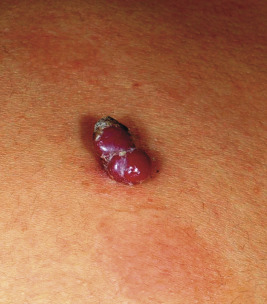
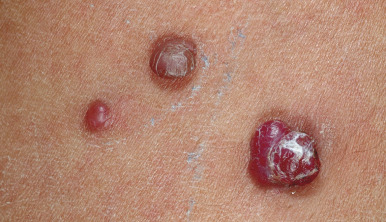
The traditional therapy for PG is simple shave excision followed by treatment of the base with electrodesiccation, which achieves hemostasis and seems to help prevent recurrence. PDL therapy may be useful for the treatment of smaller lesions, and the continuous wave/pulsed carbon dioxide (CO 2 ) laser has also been demonstrated effective. Topical imiquimod cream, an immune response modifier, may be helpful in some patients.
Bacillary Angiomatosis
Bacillary angiomatosis (BA) is an exanthem characterized by cutaneous vascular lesions in association with Bartonella (previously Rochalimaea ) infections. Bartonella species also cause cat-scratch disease, prolonged fever, hepatosplenic disease, ocular manifestations (including Parinaud oculoglandular syndrome), encephalopathy, hemolytic anemia, osteomyelitis, endocarditis, glomerulonephritis and pulmonary disease. BA occurs primarily in immunocompromised individuals and was originally described (and once seen quite commonly) in patients with acquired immunodeficiency syndrome (AIDS). It has been rarely documented in immunocompetent children. BA is caused by B. henselae and B. quintana , and transmission is via the body louse, a cat scratch, or cat fleas. The lesions may involve various tissues, including brain, bone, lymph nodes, GI tract, respiratory tract, and bone marrow. Osseous lesions are most commonly seen with B. quintana infection, and visceral involvement is caused almost solely by B. henselae. However, skin lesions are the most common manifestation, may be caused by both species, and present as disseminated red to purple papules generally no larger than 1 to 2 cm in diameter. The clinical differential diagnosis may include PG and Kaposi sarcoma. Occasionally, BA may present with ulcerative lesions. Liver peliosis is a similar condition affecting the liver and lymph nodes and usually caused by B. quintana.
The diagnosis of BA is confirmed by tissue biopsy with histologic examination, which reveals vascular proliferation and plump endothelial cells with the infecting bacilli identified on Warthin–Starry stain. The possibility of human immunodeficiency virus (HIV) infection should be considered if BA is diagnosed. The disorder usually responds to antibiotic therapy with erythromycin, doxycycline, trimethoprim-sulfamethoxazole, or rifampin.
Glomus Tumor
Glomus tumor (glomangioma, glomuvenous malformation) is a benign vascular lesion that usually presents as a blue papule or nodule. It represents a relatively uncommon hamartoma of the glomus body, which is a temperature-regulating AV shunt that bypasses the usual capillary bed of the dermis. The glomus cell, which proliferates in this disorder, is a modified smooth-muscle cell. Rarely seen in infants, these lesions may be solitary or multiple and occur in children as well as adults. Occasionally, nodular or plaque-like lesions may also occur.
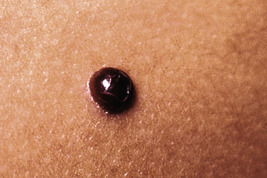
Solitary glomus tumors, which represent 90% of all lesions, do not appear to have a familial tendency. They are characterized by the clinical triad of paroxysmal pain, local tenderness, and cold sensitivity. Solitary lesions present as a blue-red nodule ( Fig. 12-52 ) from 1 mm to several centimeters in size. They most often appear on the upper extremities, particularly the nailbeds, and occasionally on the lower extremities, head, neck, or penis. Although the etiology is unknown, some lesions appear to be associated with previous trauma. The differential diagnosis of a solitary glomus tumor includes VM, blue nevus, melanoma, dermatofibroma, and leiomyoma. If occurring in an infant, hemangioma of infancy may also be in the differential.
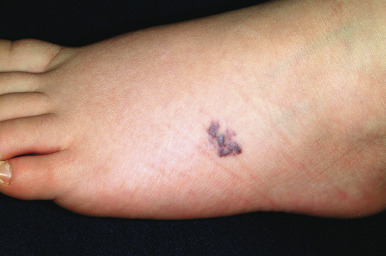
In contrast to the solitary type, multiple glomus tumors (often referred to as glomuvenous malformations ) are often dominantly transmitted, may be painful or painless, and vary from a few lesions to several hundred. The familial nature may not be obvious, because family members may have small lesions and never seek treatment. They are relatively more common in children than adults, and although they can occur anywhere on the cutaneous surface, the majority involve the lower extremities. They may be regionally distributed or generalized, and segmental patterns of presentation have been reported. Affected individuals usually have truncating mutations in the glomulin ( GLMN ) gene. Multiple glomus tumors appear as blue-purple, flat to dome-shaped papules, plaques, and nodules ( Fig. 12-53 ) that vary from a few millimeters to several centimeters in size. The differential diagnosis of multiple glomus tumors includes leiomyomas, diffuse hemangiomatosis, and blue rubber-bleb nevi. Multiple glomus tumors involving the fingers and toes have been reported in some individuals with neurofibromatosis type 1.
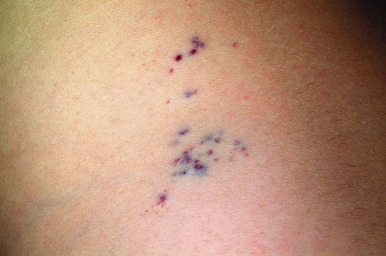
A congenital variant of glomus tumor has been described and is characterized by a blue-red, nodular plaque that may be painful and tends to grow proportionate with the child’s growth. Congenital facial plaque-like glomus tumors may mimic VMs and can be quite disfiguring.
Treatment of glomus tumors consists primarily of surgical excision, although this is not a feasible option for patients with multiple lesions and recurrence tends to be common. Sclerotherapy has been used with some success, as have CO 2 lasers. Subungual glomus tumors are usually treated successfully with periungual or transungual surgical excision, although residual nail deformities may result.
Hemangiopericytoma
Hemangiopericytoma is a rare tumor that occurs in both an adult and a childhood form, although children account for fewer than 10% of all cases. Congenital lesions are occasionally observed. This tumor arises from pericytes, which are smooth-muscle cells that surround capillaries and postcapillary venules. Skin, subcutaneous, and muscular tissues maybe involved, and any part of the body can be affected, the most common location being the lower extremities. Some experts question whether a majority of lesions previously called hemangiopericytomas actually represent solitary fibrous tumors (originating from fibroblasts).
Hemangiopericytoma may present in a variety of fashions without a distinctive or pathognomonic appearance. It often presents as a deep soft-tissue mass with slow growth. It may be flesh-colored or have a blue-red hue. Multicentric lesions have been described in infants and tend to behave in a fashion similar to their solitary counterparts. The diagnosis of hemangiopericytoma is based on the cellular and architectural features on tissue histology.
The prognosis of this tumor in childhood is variable. It appears that children younger than 1 year of age (in which case, it is termed infantile hemangiopericytoma or the congenital/infantile form of hemangiopericytoma ) have a better prognosis with a high response to chemotherapy. In children older than 1 year of age, the tumor behaves more similarly to those in adults and may be more aggressive. Treatment consists of complete surgical resection when feasible, as well as radiotherapy and rarely chemotherapy.
Angiolymphoid Hyperplasia with Eosinophilia
Angiolymphoid hyperplasia with eosinophilia (ALHE) is an uncommon vascular proliferation disorder occurring most commonly in young adult women and only rarely in children. It presents as a subcutaneous mass of the head and neck region, especially around the ears or on the scalp. Regional lymph node enlargement and eosinophilia may be present, and treatment is usually by surgical excision, although observation, cryotherapy, laser ablation, steroids, radiation, and chemotherapy have been reported.
Kimura disease is a closely related, yet distinct, chronic inflammatory disorder that occurs primarily in young Asian males. It is characterized by the triad of painless subcutaneous nodules in the head or neck region, blood and tissue eosinophilia, and elevated serum immunoglobulin (Ig)E levels. Clinically, the lesions of Kimura disease present as solitary or multiple, purple-red papules, nodules, or deep swellings. The parotid and submandibular glands may be involved, as may the oral mucosa, scalp, ears, or orbit. Unilateral cervical lymphadenopathy is commonly present. Laboratory evaluation consistently reveals eosinophilia and increased IgE. Pediatric Kimura disease may be associated nephritic syndrome, urticaria, and eczema.
ALHE and Kimura disease may be confused with a variety of other entities, most commonly malignancies such as lymphoma, salivary gland tumors, or histiocytosis. These disorders histologically reveal vascular proliferation with eosinophils and mast cells, and their etiology remains unclear. Treatment options for Kimura disease include surgical excision (although recurrence is common), steroids, chemotherapy, and radiation.
Vascular Malformations and Malformation Syndromes
Salmon Patch
The salmon patch (nevus simplex) is the most common vascular lesion of infancy. It occurs in 30% to 40% of all newborns and appears as a flat, dull pink, macular lesion on the posterior neck and scalp ( Fig. 12-54 ), glabella ( Fig. 12-55 ), forehead, upper eyelids, and occasionally the nose or nasolabial regions. When seen on the nape of the neck, it is commonly referred to as a stork bite and when on the forehead/glabella, as the angel kiss. Although the salmon patch represents the most common form of capillary malformation, it is felt by many to be a form of persistent fetal circulation rather than a true malformation. Although older nomenclature may include salmon patch under the category of PWS, this terminology should not be used because these lesions have distinct natural histories and differing significance in terms of potential syndrome associations. Salmon patches are usually isolated lesions without other associated findings. Nuchal or occipital stains may occasionally develop an associated overlying dermatitis ( Fig. 12-56 ), which may respond to topical corticosteroids or PDL treatment, if necessary.
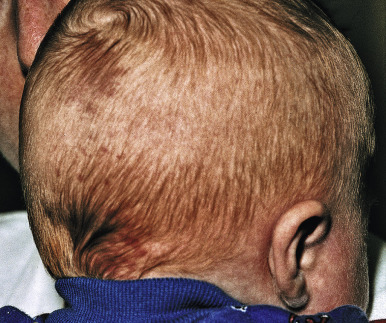
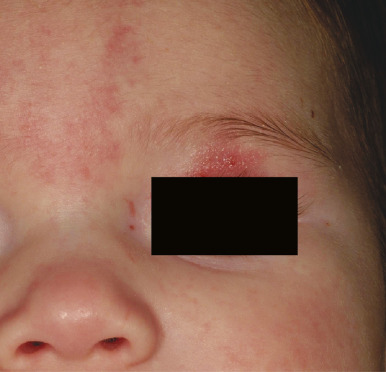
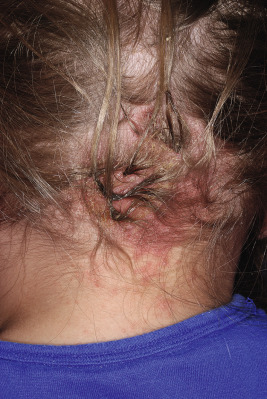
No treatment is usually necessary for salmon patches, because 95% of facial lesions fade within the first 1 to 2 years of life. Lesions on the posterior neck or scalp (also known as Unna nevus ) may fade, although some of these persist indefinitely. Since they are usually covered by hair, these lesions do not typically pose a cosmetic problem. Parents should be educated regarding the common finding of “reappearance” or accentuation of facial lesions during episodes of crying, breath-holding, straining with defecation, or physical exertion.
Nevus Flammeus/Port wine Stain
Nevus flammeus, or PWS, is a congenital capillary malformation that may occur as an isolated lesion or in association with a variety of syndromes. These lesions present as macular (nonpalpable) stains with a pink ( Fig. 12-57 ) to dark red ( Fig. 12-58 ) color. Although an early PWS may be indistinguishable from an IH, these lesions are usually distinguished by their congenital presence and their static nature without the rapid proliferation and thickening that characterizes hemangiomas during the first months of life. PWS may darken progressively over many years, and occasional lesions develop secondary proliferative (PG-like) vascular blebs on their surface (see Fig. 12-51 ). They may also become somewhat thickened and raised later in life. PWSs are often, but not always, unilateral and the most common site of involvement is the face, although they may occur on any cutaneous surface.
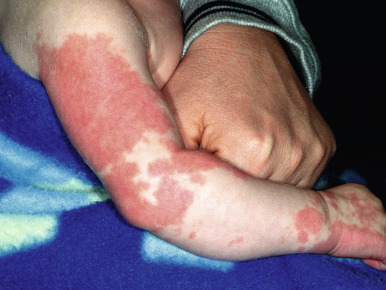
The significance of PWS is two-fold: the potential cosmetic impact of the lesion (especially facial PWS) on the developing child, and the potential for syndrome associations. Box 12-4 lists the various congenital syndromes that may have PWS as a component. Several of these will be discussed later.
Sturge–Weber syndrome
Klippel–Trénaunay syndrome
Parkes Weber syndrome
Phakomatosis pigmentovascularis
Proteus syndrome
CLOVES syndrome
Macrocephaly-capillary malformation (M-CM) syndrome
Capillary malformation-arteriovenous malformation (CM-AVM) syndrome
Cobb syndrome
Bannayan–Riley–Ruvalcaba syndrome
Beckwith–Wiedemann syndrome
Von Hippel–Lindau disease
Rubinstein–Taybi syndrome
Wyburn–Mason syndrome
Roberts syndrome
Coat disease
CLOVES, Congenital lipomatous overgrowth, vascular malformations, epidermal nevi, and scoliosis/skeletal and spinal anomalies.
PWS lesions show little tendency toward spontaneous improvement or involution, and traditional therapy was limited to the use of tinted “cover-up” cosmetics (such as Covermark or Dermablend). Laser therapy has revolutionized the treatment of these lesions, and the flashlamp-pumped tunable PDL is the most accepted laser for PWS treatment. PDL allows the targeting of short bursts of energy at intravascular hemoglobin (because of the specific wavelength of the emitted light) within the lesional vessels while sparing other tissue components and thus allowing for precise therapy. The absorbed light is converted into thermal energy, which leads to the coagulation of blood vessels. Treatment of PWS with PDL is usually performed in conjunction with local or general anesthesia, depending on the size and location of the lesion and the age of the patient. Some smaller lesions can be treated in the clinic setting without the need for anesthesia. PDL therapy is usually performed over several sessions separated in time by 6 to 8 weeks and can be quite effective in lightening these lesions, thereby minimizing their cosmetic and psychosocial significance. Although there may be psychological benefits of PDL therapy of facial PWS early in life, studies evaluating efficacy as a function of patient age have yielded mixed results. Some authors have reported optimal treatment response in patients younger than 1 year of age, whereas others have found no evidence that treatment during early childhood is more effective than treatment at a later age. In general, lesions over bony areas of the face (i.e., the forehead), lateral cheeks, chest, and proximal arms seem to respond best to PDL therapy, whereas those over the midface and distal extremities respond less well. PWS of the central face may not respond as well, given the deeper vessels that may escape the effect of the PDL. When PDL is being considered for PWS, it is important for the patient/parents to understand that complete clearance is rare, and 20% to 30% of PWSs are resistant to the treatment.
Sturge–Weber Syndrome
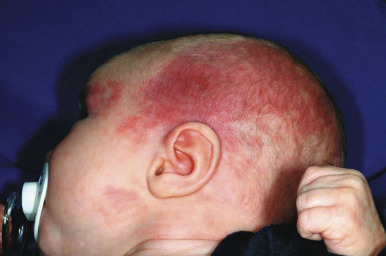
Sturge–Weber syndrome (SWS; encephalofacial or encephalotrigeminal angiomatosis) is a neuroectodermal syndrome characterized by a PWS in the distribution of the first (ophthalmic) branch of the trigeminal nerve (V1) in association with leptomeningeal angiomatosis (presenting usually with seizures) and glaucoma. There are rare reports of patients with classic brain and ophthalmic findings of SWS in the absence of a facial PWS. A somatic activating mutation in GNAQ (specifically, a single-nucleotide variant c.548G>A, p.Arg183Gln) was recently identified as the etiology behind SWS and has also been found in a significant number of individuals with nonsyndromic PWS.
Patients with SWS have a PWS in a V1 distribution ( Fig. 12-59 ) and may also have multidermatomal or more extensive cutaneous involvement. Overall it appears that around 8% of patients with a trigeminal PWS have associated glaucoma and/or seizures. Involvement of the V1 dermatome seems to be consistent in all patients. Involvement of the other trigeminal dermatomes may be useful in predicting the risk of SWS. In one study, involvement of V1, V2, and V3 was associated with an increased incidence of SWS, as was bilateral trigeminal involvement. Another study comparing unilateral to bilateral facial PWS found a higher incidence of glaucoma and leptomeningeal angiomatosis with bilateral involvement and greater risk of CNS involvement when there was complete (vs. incomplete) involvement of V1. Although the exact innervation of the lower eyelid is controversial (V1 vs. V2), it seems that involvement of both upper and lower eyelids portends a significantly higher risk of SWS. When PWS lesions are unilateral, there is usually a sharp demarcation at the midline.
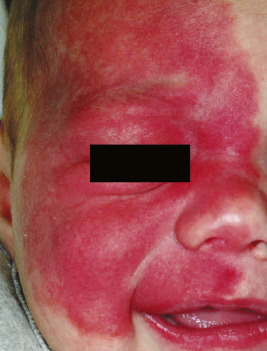
CNS disease is another component of SWS. Seizures are the most common CNS feature and often have their onset during the first year of life. The seizures of SWS may be difficult to control, and both early onset and increased seizure intensity are associated with future developmental and cognitive delay. Headaches (including migraines), stroke-like episodes, focal neurologic impairments, cognitive deficits, and emotional and behavioral problems including depression, violent behavior, and self-inflicted injury are also more common in SWS. Low self-esteem is common in relation to the facial PWS.
Leptomeningeal angiomatosis is a classic component of the syndrome, and lesions are commonly ipsilateral to the cutaneous vascular stain. Cerebral atrophy is a common radiologic finding, as is enlargement of the choroid plexus and venous abnormalities. MRI is the modality of choice for identifying these changes, although CT scans are better at detecting the classic cortical calcifications, which are also seen. These calcifications follow the convolutions of the cerebral cortex and are characterized by double-contoured parallel streaks of calcification (“tram lines”). Intracerebral calcifications may be visible during early infancy and can often help confirm the diagnosis before other characteristic features are present. Diffusion MRI, postcontrast fluid-attenuated inversion recovery (FLAIR) imaging, and high-resolution blood oxygen level-dependent (BOLD) magnetic resonance venography may be useful in the imaging of suspected SWS. Electroencephalography (EEG) is a good option for assessing abnormal brain activity in young children and may be useful in identifying patients at risk for future neurologic symptoms. However, a concerning EEG should always be followed by appropriate radiographic imaging.
Ocular involvement occurs in around 60% of patients with SWS. Glaucoma is the most common ocular finding, and it may present at any time between birth and the fourth decade. It may be unilateral or bilateral, with the latter being more common in patients with bilateral facial PWS. Vascular malformations of the eye in patients with SWS may involve the conjunctiva, episclera, choroid, and retina. Other eye findings include nevus of Ota, buphthalmos, and blindness.
Care of the child with SWS should be multidisciplinary. Dermatologic, neurologic, and ophthalmologic follow-up care is indicated, and the primary care provider must provide anticipatory guidance and support. Other specialty services may become necessary, including plastic surgery, neurosurgery, interventional radiology, and physical and occupational therapy. Referral to support organizations is useful. The Sturge–Weber Foundation ( www.sturge-weber.org ) provides education and support to individuals with this condition and other disorders associated with PWS.
The primary aim of treatment in SWS is the minimization or elimination of seizure activity. Although the primary management for seizures is with pharmacologic agents, surgical therapy may become necessary. Visually guided lobectomy with excision of the angiomatous cortex is considered the primary surgical approach in a patient with focal lesions. Hemispherectomy is often advised for patients with intractable seizures and unihemispheric involvement. This radical therapy is often successful, with decreased seizure activity and in some patients, cognitive and behavioral improvement. Presymptomatic therapy with low-dose aspirin has been advocated by some for young children before the onset of seizures or strokes in an effort to decrease the incidence of these complications. The modified Atkins diet and ketogenic diet have been used in some patients to help control seizures.
Phakomatosis Pigmentovascularis
Phakomatosis pigmentovascularis ( PPV ) is a term used to describe the association of a nevus flammeus (PWS) with a pigmented nevus or in some cases, a nevus anemicus. PPV has traditionally been classified into four types. More recently, a fifth type of PPV has been proposed as the concomitant findings of cutis marmorata telangiectatica congenita (CMTC) and mongolian spots. The classification of PPV is shown in Table 12-5 , and the cause of this disorder remains unclear. Nevus anemicus is a distinct vascular birthmark characterized by blanching of cutaneous blood vessels, hence presenting as a “white” (actually vasoconstricted) patch of skin ( Fig. 12-60 ) that becomes unnoticeable when the surrounding skin is blanched with a glass slide (diascopy). These lesions may occur as part of PPV (types II, III, and IV) but may also be seen as sporadic lesions.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


