Endocrinologic diseases often display cutaneous features that may provide diagnostic clues, and patients with these disorders may be more susceptible to a variety of mucocutaneous problems. Some of these conditions and their therapy, where applicable, are reviewed here. A thorough discussion of all endocrine disorders and their treatment is beyond the scope of this chapter.
Thyroid Disorders
Patients with hyperthyroidism or hypothyroidism may show a variety of skin findings. In some cases associated hair or nail defects may also be seen. Some of these findings are related to the imbalance in thyroid hormone, which may play a role in skin homeostasis via its influence on proteoglycan synthesis, epidermal differentiation, hair formation, and sebum production.
Hyperthyroidism
Box 23-1 summarizes the various cutaneous changes that may be seen in hyperthyroidism. This disorder is most often caused by an autoimmune thyroid disease, Graves disease. It may also be associated with a hyperfunctioning thyroid nodule, thyroid multinodular goiters, non-Graves thyroiditis, excessive thyroxine intake, or hypersecretion of thyroid-stimulating hormone (TSH). Cutaneous features occur most commonly in association with Graves disease, and these manifestations may more often be the result of autoimmune mediation rather than direct effects of thyroid hormones.
Warm, velvety, moist, erythematous, smooth skin
Thin, fine hair; diffuse nonscarring alopecia
Hyperpigmentation (especially of palmar creases, soles, gingivae, buccal mucosa)
Facial flushing
Palmar erythema
Increased sweating
Nail changes (onycholysis, curvature, koilonychia, clubbing, yellow nails)
Pretibial myxedema
Periorbital edema
Chronic urticaria
Generalized pruritus
Thyroid acropachy (clubbing)
Increased incidence of vitiligo, alopecia areata
Gynecomastia (in men)
The clinical features of hyperthyroidism include nervousness, emotional lability, tachycardia and palpitation, heat intolerance, weakness, fatigue, tremors, hyperactivity, increased appetite, weight loss, increased systolic and pulse pressure, accelerated growth, sleep disturbances, school problems, vomiting, diarrhea and occasionally exophthalmos. An enlarged thyroid gland is often present. Specific findings that suggest Graves disease include thyroid ophthalmopathy, pretibial myxedema (PTM), and acropachy. The ophthalmopathy is secondary to periorbital deposition of glycosaminoglycans and may present with proptosis, ocular paralysis, or lid lag.
Pretibial Myxedema
Pretibial myxedema (PTM; also known as thyroid dermopathy ) is manifested as plaques on the anterior tibial surfaces and is related to localized accumulation of acid mucopolysaccharides. Other locations of involvement are occasionally reported, including the dorsal feet. The majority of patients with PTM have ophthalmopathy. The onset of PTM usually follows the diagnosis of hyperthyroidism. Examination reveals nonpitting edema, nodules, and plaques with pink to yellow-brown discoloration. The overlying epidermis is thin with a waxy and sometimes translucent quality. Hypertrichosis with dilated hair follicles and a “peau d’orange” appearance may be noted. Polypoid or elephantiasis forms occur less commonly. Although the pretibial surfaces are most commonly involved, other areas including the face, scalp, upper extremities, and trunk may reveal similar changes. The differential diagnosis may include cellulitis, trauma, erythema nodosum, or other mucinoses, and PTM can be confirmed by tissue examination of skin-biopsy material in conjunction with laboratory findings. Histologic evaluation may reveal mucin deposition and reduced elastic fibers.
Thyroid Acropachy
Thyroid acropachy is more common in adults with hyperthyroidism, only occasionally occurs in children, and classically presents as the triad of digital clubbing, soft-tissue swelling of the hands and feet, and periosteal changes. Examination reveals thickening of the soft tissues over the distal extremities and (secondary to diaphyseal periosteal proliferation) of the distal bones. Drumstick-like clubbing, enlargement of the hands and feet, and radiographic changes (fluffy, spiculated, or homogeneous subperiosteal thickening with new bone formation) may be seen. Associated extremity and joint pain may be reported, and nail clubbing may also be seen. Thyroid acropachy and diabetic dermopathy (DD) are indicators of more severe Graves ophthalmopathy.
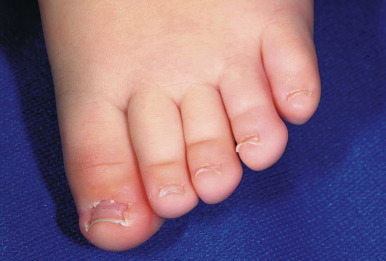
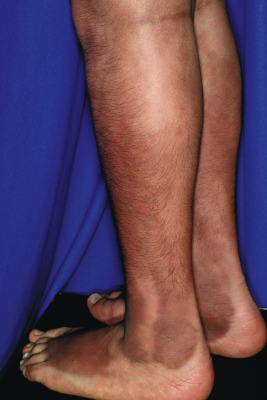
Less specific cutaneous changes in hyperthyroidism include skin that is warm, velvety, moist, and smooth. Hair may be fine and thin. Facial flushing, increased sweating (particularly of the palms and soles), and palmar erythema are common, especially in the advanced state of the disease, called thyrotoxicosis . The nails grow rapidly, are shiny, and may reveal onycholysis (separation or loosening of the nail plate from the nailbed) with distal upward curvature (Plummer nails). Koilonychia (spoon-shaped nails; Fig. 23-1 ) and clubbing may also be noted. Chronic urticaria and generalized pruritus are uncommon manifestations of thyrotoxicosis, and chronic active hyperthyroidism can be complicated by Addison disease-like hyperpigmentation. However, the hyperpigmentation of hyperthyroidism is distributed differently, primarily on the shins ( Fig. 23-2 ), posterior feet, and nailbeds. Patients with hyperthyroidism may also have an increased incidence of alopecia areata and/or vitiligo. Gynecomastia may be present in males and may be caused by increased conversion of testosterone to estradiol. Scleromyxedema, a condition marked by white-yellow papules, weight loss, monoclonal gammopathy, esophageal dysmotility, myopathy, and Raynaud phenomenon, has also been reported in patients (mainly adults) with hyperthyroidism.
Hypothyroidism
Box 23-2 summarizes the various mucocutaneous changes that may be seen in hypothyroidism. Hypothyroidism in pediatric patients may be congenital or acquired. Clinical features of congenital hypothyroidism are shown in Box 23-3 . Symptoms may be subtle in both forms of hypothyroidism, but decreased linear growth is an important indicator of both types of disease. Hypothermia, lethargy, and poor feeding are all characteristic of congenital disease, which is most often diagnosed on routine newborn screening examinations. Children with acquired hypothyroidism tend to be quiet and well behaved and as a result often receive high grades in school.
Dry, coarse, pale, cool skin
Dull, brittle hair with thinning
Cutis marmorata
Generalized myxedema (especially hands, feet, periorbital)
Carotenoderma
Nail changes (ridged, brittle, grow slowly)
Alopecia (diffuse, lateral eyebrows)
Hypertrichosis (back and shoulders usually)
Easy bruising
Ichthyosis
Eruptive or tuberous xanthomas
Livedo reticularis
Periorbital edema
Dermatitis herpetiformis (has been reported with Hashimoto thyroiditis, atrophic variant)
Protuberant lips
Macroglossia
Urticaria
Pruritus
Puffy (myxedematous) facies
Sallow complexion
Wide anterior fontanel and sutures
Macroglossia and thick lips
Hypertelorism, depressed nasal bridge
Coarse, brittle hair
Hoarse cry
Translucent (“alabaster”) ears
Umbilical hernia and abdominal distention
Heart murmur
Hypotonia and slow reflexes
Short stubby fingers and broad hands
Short lower extremities
Scalp seborrhea, purpura
Prolonged relaxation phase of tendon reflexes
Cold, mottled, or jaundiced skin
Sluggishness and inactivity
Delayed motor development, mental retardation
Lack of coordination and ataxia
Poor weight gain, stunted growth
Subnormal body temperature, poor circulation, and intolerance to cold
Delayed/defective dentition
Myxedema (some cases)
Congenital hypothyroidism may develop as a result of agenesis or dysgenesis of the thyroid gland (the most common cause); defective synthesis of thyroid hormone caused by an enzymatic defect; the presence of antithyroid antibodies in a pregnant mother; the lack of maternal iodine during pregnancy (endemic goiter); or the ingestion of antithyroid medications such as propylthiouracil or methimazole by a pregnant woman being treated for thyrotoxicosis. Iodine toxicity from iodine-containing skin antiseptic solutions has been implicated as a potential cause of transient hypothyroidism in newborn infants, although recent studies refute this association. Acquired hypothyroidism in children younger than 5 or 6 years of age may be caused by a delayed failure of thyroid remnants (with thyroid dysgenesis); by inborn defects of thyroid-hormone synthesis; by ingestion of antithyroid agents; by thyroidectomy or ablation after radiation; or by chronic thyroiditis or hypothalamic-pituitary disease. After the age of 5 or 6 years, although the same etiologies may be involved, chronic lymphocytic thyroiditis (Hashimoto thyroiditis) is the most common cause. Although iodine deficiency is the most common cause of hypothyroidism worldwide, it is uncommon in the United States.
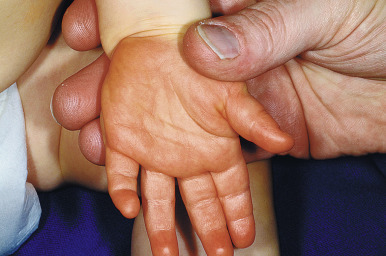
Cutaneous features of hypothyroidism are a reflection of the hypometabolic state with reduced body temperature and reflex vasoconstriction. Patients often have dry, coarse, pale, and cool skin. Cutis marmorata (physiologic mottling) may be prominent. Hypohidrosis may lead to acquired palmoplantar keratoderma. Myxedema may occur in hypothyroidism, again as a result of mucopolysaccharide deposition in skin. It most commonly occurs in the hands, feet, and periorbital locations and may also be deposited in the tongue, giving rise to macroglossia. Generalized puffiness may be present, and the skin may take on a yellow hue as a result of carotenemia ( Fig. 23-3 ). The hair is dull and brittle, and nails are ridged, brittle, and grow very slowly. Patients may have a dull, expressionless facies. Hypertrichosis of the back and shoulders may be seen, and alopecia may involve the lateral portions of the eyebrows (termed madarosis ) or occur in a more diffuse pattern. A collodion baby with concomitant congenital hypothyroidism has been reported.
Parathyroid Disorders
Disorders of the parathyroid glands (hyperparathyroidism and hypoparathyroidism) may affect the skin in various ways and often present with cutaneous features that can assist the primary or consulting physician in diagnosis and management. Although primary parathyroid disease is uncommon in children, these glands play a major role in the regulation of calcium and phosphorus metabolism, and associated abnormalities manifest distinctive clinical patterns. Parathyroid hormone (PTH) is one of the two main calciotropic hormones (the other one being calcitriol); these hormones regulate phosphate and calcium homeostasis.
Hyperparathyroidism
Primary hyperparathyroidism, one of the least common endocrine disorders of infancy and childhood, is rarely diagnosed in children younger than 16 years of age. When seen, it is usually the result of a familial, genetically determined hyperplasia of the parathyroid, which may present as an isolated hyperparathyroidism or as the hyperparathyroidism–jaw-tumor syndrome, in which case ossifying tumors of the maxilla or mandible are present. A malignant neoplasm of the parathyroid, or an association with some other disease such as is seen in patients with multiple endocrine adenomatosis (multiple endocrine neoplasia [MEN]; see below) or chronic renal insufficiency may also result in hyperparathyroidism.
The majority of cases of hyperparathyroidism are sporadic, most often being caused by a single adenoma in the parathyroid gland. The clinical features of hyperparathyroidism include systemic effects of hypercalcemia: failure to thrive, muscular weakness, lethargy, anorexia, vomiting, fever, headache, constipation, weight loss, polydipsia, polyuria, mental retardation, metastatic calcification, and with marked hypercalcemia, stupor or death. Of these, metastatic calcification is the most common cutaneous manifestation, and in patients with sporadic hyperparathyroidism, this may be the only cutaneous finding. Hypercalcemia may also produce an ophthalmologic finding known as band keratopathy , which is the result of calcium and phosphate deposition beneath the Bowman capsule. Band keratopathy appears as a superficial corneal opacity resembling frosted or ground glass in a band-like configuration with white flecks or “holes” in the band resulting in a “Swiss cheese”-like appearance. It is not specific for hyperparathyroidism but may also be seen as a manifestation of hypercalcemia secondary to vitamin D intoxication, uremia, or sarcoidosis. It is not commonly found in patients with hyperparathyroidism when serum phosphorus levels are low and glomerular function is maintained.
Patients with chronic renal failure may experience several types of cutaneous manifestations. The skin of the patient with uremia may be pruritic, dry, scaly, sallow, and hyperpigmented; the sallow appearance is partially caused by anemia, and the hyperpigmentation appears to be the result of decreased renal clearance of melanocyte-stimulating hormone (MSH). Hyperparathyroidism secondary to chronic renal failure results from impaired synthesis of 1,25-dihydroxyvitamin D 3 , which leads to hypocalcemia from impaired intestinal calcium absorption and ultimately, increased levels of PTH. Hyperphosphatemia may result in a high serum calcium phosphate product and produce secondary calcification of the skin. This calcinosis cutis (see Chapter 9 ) manifests as hard calcium deposits in skin and subcutaneous tissues, especially in periarticular locations. These lesions may resolve spontaneously with correction of the serum calcium and phosphate levels. In addition to being seen in hyperparathyroidism, it may also be noted in association with paraneoplastic hypercalcemia, milk alkali syndrome, sarcoidosis, and hypervitaminosis D.
When the calcification is more progressive and involves blood vessels, ischemic necrosis of skin and soft tissues occurs and is termed calciphylaxis . This rare (especially in children) and life-threatening condition results from vascular calcification and is most commonly reported in patients with end-stage renal disease. Clinically, it is manifested as ecchymotic or infarcted areas of skin, bullous lesions, and plaques of calcinosis with periodic extrusion of calcium. Lesions of pediatric calciphylaxis are most commonly noted on the upper and lower extremities. They are very painful, and mortality related to gangrene and sepsis is high. Extensive calcification in the heart and lungs may result in cardiorespiratory failure. Parathyroidectomy is often, but not always, useful in this setting. Histologically, calciphylaxis shows calcification of the walls of small and medium-sized blood vessels in the dermis and subcuticular regions.
The diagnosis of hyperparathyroidism is established by consistent elevations of total serum calcium above 12 mg/dL, the reduction of serum phosphorus concentrations below 4 mg/dL, and elevated levels of PTH. High alkaline-phosphatase levels usually indicate bone disease. This complication of hyperparathyroidism may be demonstrated radiographically by generalized demineralization of bones, destructive changes at the growing ends of long bones, subperiosteal erosions (particularly in the phalanges, metacarpals, and lateral portions of the clavicles), and in more advanced disease, generalized rarefaction, cysts, tumors, fractures, and deformities. Radiographs of the abdomen may reveal renal calculi or nephrocalcinosis, and ultrasonography and radioisotope scanning can confirm the diagnosis of primary hyperparathyroidism associated with an isolated parathyroid adenoma. In infants with parathyroid hyperplasia, cupping and fraying at the ends of long bones and ribs may suggest rickets, and severe demineralization and pathologic fractures are common.
Hypoparathyroidism/DiGeorge Syndrome
Hypoparathyroidism is characterized by hypocalcemia and inappropriate response of the parathyroid glands or, less often, with elevated PTH levels and lack of response to the hormone (see Pseudohypoparathyroidism section). In childhood, hypoparathyroidism may develop as a congenital idiopathic disorder but usually appears in the neonatal period, in later infancy, or during childhood or as an acute condition after inadvertent removal or damage of the parathyroid glands during thyroid surgery. Congenital hypoparathyroidism may occur alone; may be seen as an autoimmune disorder, where it may occur alone or with other endocrine disorders; or may be a hereditary condition associated with an increased familial incidence of other endocrinologic disorders (Addison disease, pernicious anemia, and Hashimoto thyroiditis), candidiasis, and/or vitiligo. When associated with hypoplasia of the thymus and immunologic defects, the condition is known as DiGeorge syndrome (see below).
Idiopathic or congenital hypoparathyroidism usually is first manifested by tetany or seizures and in 25% to 50% of patients, ectodermal defects. The skin of affected individuals is rough, dry, thick, and scaly; the hair and eyebrows are sparse; and the nails are short and thin with brittleness, crumbling, or longitudinal grooving. When hypoparathyroidism occurs during tooth development, pitting, ridging, absence of dental enamel, and absence or hypoplasia of the permanent teeth may result. Extensive calcification of skin and subcutaneous tissues has been reported in an infant with congenital primary hypoparathyroidism, although this is exceedingly rare. Other clinical manifestations include convulsions, carpopedal spasm, muscle cramps and twitching, numbness or tingling of the extremities, laryngospasm or bronchospasm, exfoliative dermatitis, mental retardation, chronic diarrhea (especially in infants), photophobia, keratoconjunctivitis, blepharospasm, and cataracts. Mucocutaneous candidiasis is seen as a complication in 15% of patients with idiopathic hypoparathyroidism. Electrocardiography may reveal prolongation of the QT interval, and head imaging may show calcifications of the basal ganglia. The combination of candidiasis, endocrinopathy, and ectodermal dysplasia has been termed autoimmune polyendocrinopathy, candidiasis and ectodermal dystrophy ( APECED ) as well as autoimmune polyglandular syndrome ( APS ) type 1 or autoimmune polyendocrinopathy syndrome type 1 and is discussed in more detail below in the section Autoimmune Polyglandular Syndromes (see also Chapter 17 ).
The cutaneous manifestations of hypoparathyroidism associated with surgical removal or injury of the parathyroid glands differ from those seen in patients with idiopathic or congenital hypoparathyroidism. These include thinning or loss of hair, the development of horizontal grooves (Beau lines) in the nails, or a complete loss of nails after episodes of tetany (these abnormalities revert to normal when hypocalcemia is controlled). Hyperpigmentation (predominantly on the face and distal extremities) may resemble melasma, pellagra, or Addison disease and also may occur in cases of postthyroidectomy hypoparathyroidism. Although cutaneous calcification has been noted, this complication is relatively uncommon. In a series of 21 patients with acquired hypoparathyroidism, the most common cutaneous manifestations were hair loss (especially axillary and pubic), coarsening of body hair, and dry skin.
DiGeorge syndrome is a T-cell deficiency disorder that develops as a result of faulty embryologic development of the thymus and the parathyroid glands (a congenital malformation of the third and fourth pharyngeal pouches and the surrounding arches). The classic triad consists of cardiac malformation, hypocalcemia, and T-cell immunodeficiency. Defects of the great vessels may include truncus arteriosus, interrupted aortic arch, double aortic arch, or aberrant subclavian artery. Oral candidiasis is an almost constant finding in patients with this disorder, and overwhelming fungal, viral, or bacterial infection usually leads to death early in infancy. Hypocalcemia and tetany may occur at an early age, and other features include chronic diarrhea, interstitial pneumonia, failure to thrive, micrognathia, hypertelorism, low-set ears, bifid uvula, shortened philtrum, bowed mouth, chronic purulent rhinitis, mental retardation, calcification of the central nervous system, and nephrocalcinosis.
DiGeorge syndrome is associated with a deletion in the long arm of chromosome 22, and is also referred to as a chromosome 22q11.2 deletion syndrome or velocardiofacial syndrome. The candidate gene for this disorder is termed TBX1 , which encodes the T-box transcription factor 1. Approximately half of patients with DiGeorge syndrome are hemizygous for 22q11, and they have occasionally been found to have overlapping deletions in the 10p13/14 boundary. These patients are at increased risk for developing psychiatric disorders, with one in four developing schizophrenia and one in six developing major depressive disorders. Other reported psychiatric morbidities include attention-deficit/hyperactivity disorder, oppositional defiant disorder, and anxiety disorders. When the immunodeficiency is severe, thymic or bone marrow transplantation should be considered.
Pseudohypoparathyroidism/Albright Hereditary Osteodystrophy
Pseudohypoparathyroidism (PHP) is a hereditary disorder in which there is decreased target tissue responsiveness in the receptor tissues, particularly the kidneys and skeletal system, to PTH (rather than a true deficiency). PHP is subclassified into types Ia, Ib, Ic, and type II (which involves a different mechanism of resistance to PTH). Albright hereditary osteodystrophy ( AHO ) refers to PHP in conjunction with a clinical constellation of physical features, including short stature, central obesity, brachydactyly, ectopic ossification, and variable degrees of mental retardation.
Pseudopseudohypoparathyroidism ( PPHP ) is a term used to describe individuals with AHO who have normal end-organ responsiveness to PTH. These patients do not develop hypocalcemia and tetany. PHP and PPHP are caused by different types of mutations in the GNAS gene, and presence of genetic imprinting may lead to quite diverse clinical phenotypes. Specifically, maternal inactivating mutations result in PHP-Ia, whereas paternal inactivating mutations result in PPHP and the disorder progressive osseous heteroplasia (POH), a disease of severe heterotopic ossifications of the subcutaneous tissues, skeletal muscles, and deep connective tissues (see Chapter 9 ).
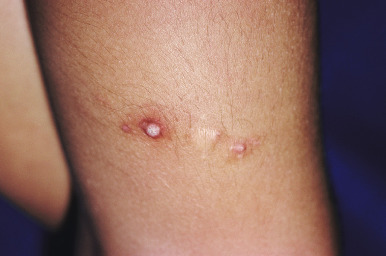
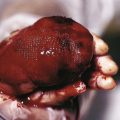
Patients with PHP have hypocalcemia, hyperphosphatemia, and elevated serum levels of PTH. Hypothyroidism secondary to TSH resistance may be seen in PHP-Ia. Ectopic calcification is common, and intracranial lesions usually involve the basal ganglia and occasionally other regions. Calcinosis cutis may occur and presents with multiple small papules, plaques, or nodules with a predilection for the scalp, hands and feet, periarticular regions, and chest wall. Soft-tissue ossification (osteoma cutis) may be present at birth or develop during infancy or childhood ( Fig. 23-4 ) and is often a presenting feature of the disease, along with hypothyroidism. The subcutaneous calcifications or ossifications may occasionally present very early in life (even by 2 weeks of age) and in those patients may be vital to early recognition and diagnosis of PHP. Dermal or subcutaneous hypoplasia may occasionally be noted in areas of cutaneous calcification.
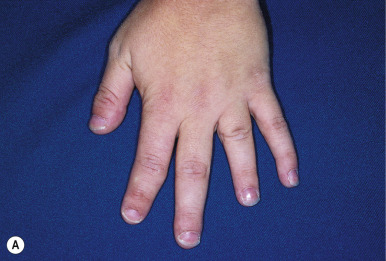
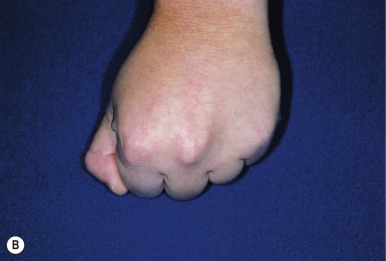
The characteristic features of AHO include short stature, obesity, and characteristic facial features, including round face, flat nasal bridge, and a short neck. Brachymetaphalangism refers to shortening of the fourth and fifth metacarpals and may be recognized by knuckle dimples when the patient makes a clenched fist. In addition, the fourth and fifth fingers and toes may appear shortened ( Fig. 23-5 ). Plain radiography may confirm this feature when the clinical findings are subtle. Mental retardation may be present and may be less common with aggressive and early treatment for the hypocalcemia.
Disorders of the Adrenal Glands
Adrenal gland dysfunction may result in a variety of systemic effects with various cutaneous manifestations. Those of particular interest to the pediatrician, dermatologist, and pediatric dermatologist are Addison disease, Cushing syndrome (CS), and the adrenogenital syndrome (discussed under Disorders of Androgen Excess).
Addison Disease
Addison disease (primary adrenal insufficiency) is caused by the absence of glucocorticoids and mineralocorticoids despite an increased concentration of adrenocorticotropic hormone (ACTH), and characterized by weakness, anorexia, weight loss, hypotension, decreased serum sodium and chloride, increased serum potassium, hypoglycemia, and hyperpigmentation of the skin and mucous membranes. Sporadic and recurrent “flu-like” episodes may provide a clinical clue to the diagnosis, especially in the setting of pigmentary alterations. Hyperpigmentation in Addison disease is the result of increased production of proopiomelanocortin, which is cleaved to form MSH and ACTH. This overproduction in the pituitary gland occurs as a compensatory phenomenon associated with decreased cortisol production by the adrenal glands. The hyperpigmentation of Addison disease occurs in the setting of primary adrenocortical failure as opposed to secondary adrenal insufficiency, in which case ACTH levels are low and mineralocorticoid production remains relatively intact.
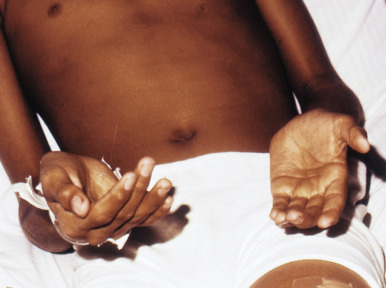
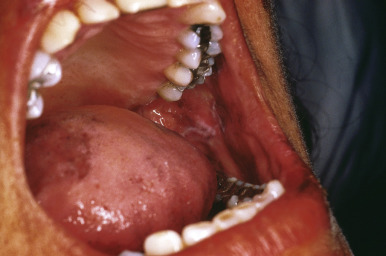
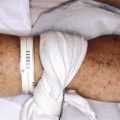
The pigmentation of Addison disease is most intense in the flexures, at sites of pressure and friction, in the creases of the palms and soles ( Fig. 23-6 ), in the nails, in sun-exposed areas, and in normally hyperpigmented areas such as the genitalia and areolae. Pigmentation of the conjunctivae and vaginal mucous membranes is common, and pigmentary changes of the oral mucosae ( Fig. 23-7 ) include spotty or streaked blue-black to brown hyperpigmentation of the gingivae, tongue, hard palate, and buccal mucosa. In addition, increased pigmentation may be noted in existing nevi. The pigmentation may in some children be quite diffuse. Labial pigmentation and longitudinal pigmentary streaks of the fingernails similar to those in Laugier–Hunziker syndrome have been observed. Because the pigmentation may in some cases be subtle, comparison of the patient to other family members may be useful in highlighting the clinical findings. In one series of 18 pediatric patients with primary adrenal insufficiency at one institution, 12 (67%) exhibited cutaneous hyperpigmentation. Primary adrenal insufficiency without hyperpigmentation has been reported and may result in a delay in the diagnosis of Addison disease. Loss of body hair may be another cutaneous finding in this disorder.
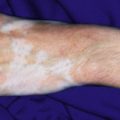
The diagnosis of chronic adrenocortical insufficiency is suggested by the clinical features and confirmed by serum electrolyte studies and cortisol level determinations after stimulation by ACTH (the ACTH-stimulation test). A morning serum-cortisol (“ am cortisol”) level is a convenient and simple test but may be insensitive as a screening tool. There are multiple potential causes of Addison disease, including adrenal dysgenesis (which may be related to a variety of gene mutations), diseases resulting in adrenal destruction, or impaired steroidogenesis (disorders of cholesterol or steroid biosynthesis, including several forms of congenital adrenal hypoplasia). Although the majority of cases of Addison disease in the past century were attributed to tuberculosis, autoimmune disease currently accounts for most cases presenting outside of the newborn period. APS (autoimmune polyendocrinopathy syndrome) types 1 and 2 may both present with this disorder as one component. Other nonautoimmune causes include infection, metabolic and infiltrative or metastatic diseases, and drug-induced damage. The incidence of Addison disease is elevated in vitiligo probands and their first-degree relatives. When these two disorders occur concurrently, patients may have a striking presentation of hypopigmentation and hyperpigmentation.
Cushing Syndrome
Cushing syndrome (CS) is a rare disorder caused by long-term glucocorticoid excess, which may be caused by a variety of different etiologies. It is divided into ACTH-dependent types (including pituitary-dependent Cushing disease, ectopic ACTH syndrome, and adrenal hyperplasia) and non-ACTH-dependent types (including adrenal adenoma, adrenal carcinoma, and adrenal hyperplasia). The most common form is pituitary-dependent bilateral adrenal hyperplasia, termed Cushing disease. Endogenous CS is fairly uncommon in children, and the lack of classic features of hypercortisolism in pediatric patients may delay diagnosis and treatment. In all patients with CS, there is loss of diurnal variation of ACTH and cortisol secretion, which leads to sustained hypercortisolism. Growth retardation to complete linear growth arrest is the hallmark of the disease in children and growing adolescents. CS may also result from the systemic administration of exogenous glucocorticoids (including oral, parenteral or rarely, topical) or ACTH, and should be suspected by the findings of suppressed ACTH and cortisol with no response to corticotropin-releasing hormone (CRH) or ACTH, respectively. CS has occurred after intralesional corticosteroid injections for keloids in a child. The authors have observed CS in a few children after topical application of ultrapotent corticosteroids for extensive alopecia areata/totalis and severe atopic dermatitis.
The clinical findings in CS are multiple and usually suggest the presence of hypercortisolism. Noncutaneous signs and symptoms include truncal obesity, marked diminution of the linear growth rate, diabetes mellitus or glucose intolerance, gonadal dysfunction, hypertension, muscle weakness, fatigue, mood disorders, sleep disturbances, menstrual irregularities, osteoporosis, delayed or accelerated bone age, edema, polydipsia, polyuria, and fungal infections. The typical growth chart in a child with CS reveals a severely diminished linear growth curve, with continued weight gain across percentiles. This is in distinction to the growth chart in a child with exogenous obesity, which reveals increasing linear growth.
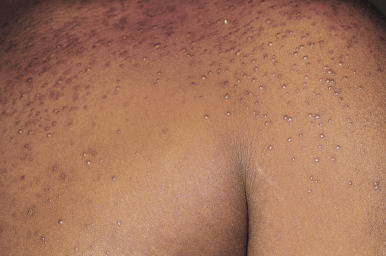
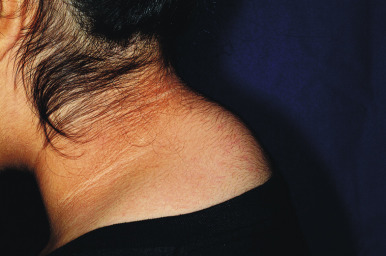
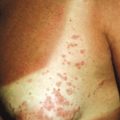
The cutaneous features of CS are listed in Box 23-4 . Addison disease-like pigmentation (particularly on the face and neck) has been noted in 6% to 10% of patients and is seen primarily in the ACTH-dependent forms of the disease. Other skin findings include a characteristic plethoric “moon” facies with telangiectasias over the cheeks; increased fine lanugo hair on the face and extremities; purplish striae (stretch marks, see below) at points of tension such as the lower abdomen, flanks, thighs, buttocks, upper arms, and breasts; fragility of dermal blood vessels with an increased tendency toward bruising at sites of minimal trauma; poor wound healing; and steroid acne. The latter usually presents as red papules or small pustules distributed primarily on the upper trunk ( Fig. 23-8 ), arms, neck, and to a lesser degree, the face. There is a tendency to develop cutaneous fungal infections (i.e., tinea corporis, onychomycosis, candidiasis, pityriasis versicolor), and disseminated mycobacterial infection has been reported. Patients with CS classically have fatty deposits over the back of the neck, termed the buffalo hump ( Fig. 23-9 ).
Facial plethora and telangiectasias
Hirsutism, fine lanugo hair growth
Violaceous striae (especially over the abdomen, flanks, and upper arms)
Acne
Bruising
Poor wound healing
Skin atrophy
Thin, translucent skin
Hyperpigmentation (seen only in ACTH-dependent form)
Acanthosis nigricans
Frequent fungal infections (i.e., tinea corporis, pityriasis versicolor, candidiasis)
Male-pattern alopecia (in females)
ACTH, Adrenocorticotropic hormone.
CS is diagnosed based on suspicious clinical findings and the results of laboratory testing. A 24-hour urinary free-cortisol test is a widely used assay for diagnosing hypercortisolism and has a very high specificity and sensitivity. Other diagnostic studies include plasma-cortisol measurement (limited by diurnal variation), low-dose dexamethasone suppression test (excellent sensitivity but requires overnight hospitalization), high-dose dexamethasone suppression test, ACTH measurement (useful for discriminating between ACTH-dependent and ACTH-independent forms), late night salivary cortisol measurement, inferior petrosal sinus sampling, and the CRH stimulation test. Radiographic imaging may be useful for assessing for pituitary or adrenal tumors. The treatment options for CS depend on the etiology and may include transsphenoidal pituitary surgery, external pituitary radiotherapy, excisional surgery (adrenal tumors), and mitotane (an adrenocytolytic agent). With the exception of striae, the cutaneous effects of CS most often completely heal after successful therapy. After treatment, glucocorticoid replacement and stress dosing of cortisol are often required.
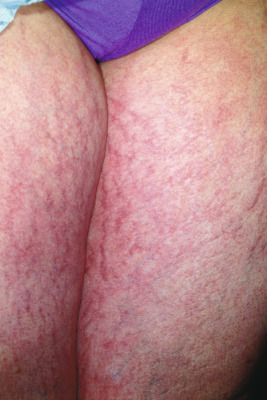
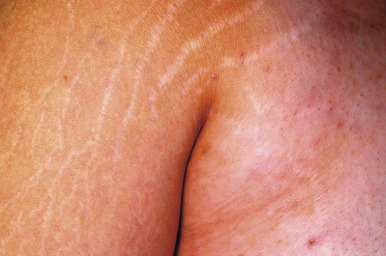
Striae distensae (stretch marks), mentioned earlier, are linear depressions of the skin that are initially pink ( Fig. 23-10 ) or purple and later become more flesh-colored, translucent, and atrophic ( Fig. 23-11 ). They are most commonly seen in areas subject to stretching such as the lower back, buttocks, thighs, breasts, abdomen, and shoulders. Striae may develop physiologically in up to 35% of girls and 15% of boys between the ages of 9 and 16 years. Causes of striae include stretching exercises, rapid growth, obesity, adolescence, pregnancy, CS, and prolonged use of systemic or potent topical corticosteroids. They may be seen in patients with anorexia nervosa, mainly the restrictive (vs. the bulimic) form. They can occasionally be mistaken for nonaccidental injury or physical abuse. Although the precise cause of striae is unknown, their formation appears to be related to stress-induced rupture of connective tissue, alteration of collagen and elastin, and dermal scarring in which glucocorticoids suppress fibroblastic activity and newly synthesized collagen fills the gaps between ruptured collagen fibers. Elastic fibers are fine in early lesions and thickened in older lesions of striae. Treatment of striae is challenging, and most therapies are generally unsatisfactory. Many of the lesions that occur during adolescence tend to become less noticeable with time. It has been suggested that topical tretinoin cream may be helpful for some patients, although the results are mixed. Other topical agents reportedly effective in some patients include glycolic and trichloroacetic acid peels and topical hyaluronic acid preparations. The flashlamp-pumped pulsed-dye laser has also been used for treating these lesions, but data suggest that this modality should be reserved for patients with the more fair skin phenotypes (types II to IV). Other treatment modalities that have been reported for striae include intense pulsed light, excimer laser, copper-bromide laser, fractional photothermolysis, and microdermabrasion.
Disorders of Androgen Excess
Patients with androgen excess may initially present with a variety of skin-related conditions, including hirsutism, acne, and alopecia. Hyperandrogenism may be related to benign or malignant ovarian, testicular, or adrenal tumors; functional overproduction of androgens by the adrenal glands or ovaries; or exogenous androgens. The clinical findings of hyperandrogenism may vary depending upon the pubertal development of the child, and these are summarized in Box 23-5 . The two disorders to be discussed in this section are congenital adrenal hyperplasia (CAH) and polycystic ovary syndrome (PCOS).
Prepubertal
Precocious sexual development:
Sex appropriate (males)
Sex inappropriate (females)
Rapid linear growth
Accelerated bone age
Axillary and pubic hair development
Acne
Hirsutism
Neonate:
Hyperpigmentation of the genitalia
Ambiguous genitalia
Postpubertal
Acne
Hirsutism
Androgenetic alopecia
Amenorrhea or irregular menses
Infertility
Other virilizing signs (deep voice, increased muscle mass, male habitus)
Early cardiovascular disease
A brief mention of exogenous androgen excess is in order, in light of the expanding US market for testosterone-replacement therapies. These products are the standard of care for androgen deficiency and are available as depot injectables, subcutaneously implanted pellets, transdermal patches, topical gels, and buccal tablets. Inadvertent drug transfer to spouses and/or children (secondary exposure) may occur in association with use of the topical agents and may present with precocious puberty in young children or virilization in exposed adult females. Although skin-to-skin contact is presumed to be the primary mode of transfer, spread via shared linens may also be possible. These reports highlight the importance of adequate education in contact precautions when topical androgens are prescribed, as well as vigilance on the part of pediatric practitioners, with recognition of this potential cause for precocious puberty. The United States Food and Drug Administration (FDA) added a box warning about the risks of secondary transfer to topical testosterone product labels in 2009.
Congenital Adrenal Hyperplasia
Congenital adrenal hyperplasia ( CAH ; also known as adrenogenital syndrome) is a term used to describe a constellation of diseases characterized by impaired steroid metabolism in the adrenal cortex. The most common cause of CAH is 21-hydroxylase deficiency, and other enzymes that may be defective include 11 β-hydroxylase and 3 β-hydroxysteroid dehydrogenase. 21-Hydroxylase deficiency occurs in about one in 10,000 to one in 15,000 live births worldwide. It is inherited in an autosomal recessive fashion, and the gene ( CYP21A2 ) is located in the HLA class III region on the short arm of chromosome 6p. Owing to the defect in cortisol synthesis, the adrenal cortex in these disorders is stimulated by corticotropin, and overproduction of cortisol precursors occurs, some of which are diverted toward synthesis of sex hormones. This androgen excess leads to clinical signs of hyperandrogenism, and concomitant aldosterone deficiency may result in salt-wasting, failure to thrive, hypovolemia, and in some cases, shock.
One of the most classic presenting features of female children with CAH (21-hydroxylase deficiency) is ambiguous genitalia ( Fig. 23-12 ). Because of exposure to high systemic levels of adrenal androgens during gestation, affected girls are born with a large clitoris, rugated and partially fused labia majora, and a common urogenital sinus in place of a separate urethra and vagina. The degree of virilization is variable and may range from simple clitoromegaly to the appearance of a penile urethra. In severe forms, important features in distinguishing the virilized female genitalia from true male genitalia include the absence of testicles and the presence of normal internal sex organs. Affected boys generally have no obvious physical signs of the disorder, although subtle hyperpigmentation and/or penile enlargement may be present. Some forms of CAH, such as 17 α-hydroxylase/17,20-lyase deficiency, may result in ambiguous or female-appearing external genitalia in males, identical to the androgen insensitivity syndrome.
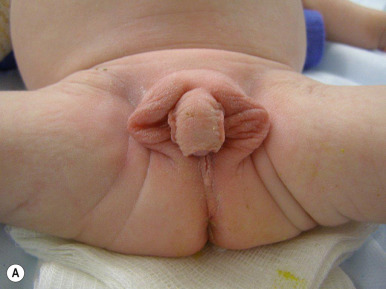
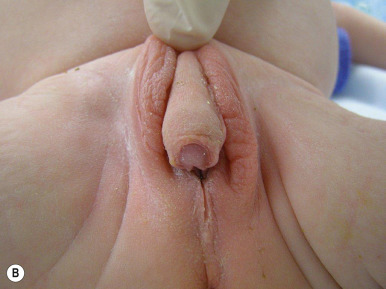
In patients who are not treated or in whom therapy is inadequate, long-term exposure to these androgens results in rapid somatic growth, advanced skeletal age, and premature development of secondary sexual characteristics, including development of pubic and axillary hair, clitoral growth, and penile growth in males. Although linear growth may be accelerated during childhood, these patients often ultimately have short stature because of premature epiphyseal closure. Centrally mediated precocious puberty may occur. Hirsutism is a common feature in patients with nonclassic (partial) 21-hydroxylase deficiency, and when menstrual irregularity and acne also occur, the presentation may simulate that of PCOS (see below). Patients with nonclassic CAH do not have ambiguous genitalia at birth and usually have premature adrenarche and advanced skeletal maturation.
Abnormalities of reproductive function are common in CAH, and infertility is often present. Affected males have fewer problems with reproductive function. Salt-wasting is seen in up to 75% of patients with classic 21-hydroxylase deficiency, owing to decreased synthesis of aldosterone. These patients are prone to hyponatremia, hyperkalemia, hypovolemia, dehydration, and shock. Hyperpigmentation, most notably of skin creases and the genitalia, may occur as an early sign of adrenal insufficiency. Classic 21-hydroxylase deficiency is diagnosed by the finding of markedly elevated levels of serum 17-hydroxyprogesterone, the main substrate for the enzyme; this is the measurement utilized in newborn-screening protocols. The cosyntropin (corticotropin)-stimulation test is useful for differentiating this disorder from other steroidogenic enzyme defects. Molecular-based genetic testing is available for CAH, and prenatal diagnosis is possible using chorionic villus and amniotic fluid cells for deoxyribonucleic acid (DNA) analysis. Fetal DNA harvested from maternal plasma has also been investigated as a noninvasive approach to prenatal diagnosis. Management for CAH consists of glucocorticoid and mineralocorticoid replacement, supplemental sodium chloride, and surgical management of ambiguous genitalia, which is generally recommended between 2 and 6 months of life. When the diagnosis is known, dexamethasone administered to the mother before the ninth week of gestation may prevent the development of genital ambiguity.
Polycystic Ovary Syndrome
Polycystic ovary syndrome (PCOS; also called Stein–Leventhal syndrome) is the most common androgen disorder of ovarian function and among the most common endocrine disorders in women. It is estimated to affect between 6% and 15% of women, depending on the diagnostic criteria utilized. Patients with PCOS have increased androgen production and disordered gonadotropin secretion, resulting in amenorrhea or severe oligomenorrhea, increased testosterone levels, and enlarged polycystic ovaries on ultrasound examination. Women with PCOS also have metabolic derangements related to insulin resistance and an increased risk of type 2 diabetes mellitus. The hormonal dysregulation seen in PCOS usually begins during adolescence. The pathophysiology of PCOS is multifactorial, and it has been found to cluster in families with a history of PCOS, non-insulin-dependent diabetes mellitus (NIDDM), cardiovascular disease, and breast cancer.
Three sets of diagnostic criteria have been utilized for adult women, including the 1990 National Institutes of Health criteria (which requires chronic oligoovulation plus clinical or biochemical signs of hyperandrogenism), the 2003 Rotterdam criteria (which requires two of three criteria: chronic oligoovulation or anovulation, clinical or biochemical signs of hyperandrogenism, and polycystic ovaries), and the criteria of the Androgen Excess Society (which defines PCOS as hyperandrogenism manifested as hirsutism and hyperandrogenemia, and ovarian dysfunction based on chronic oligoovulation or anovulation and polycystic ovaries). There are no formal diagnostic criteria in adolescents, and this may present a challenge given the potential clinical overlap between features of normal puberty and PCOS.
Hirsutism and acne vulgaris are the most common manifestations of PCOS and in fact, the leading cause of hirsutism in adolescence is PCOS (see Chapter 7 ). Hair growth occurs in androgen-dependent areas including the face, neck ( Fig. 23-13 ), chest, back, and lower abdomen. It may be less noticeable in adolescents (given their shorter duration of hyperandrogenism) and is less common in some ethnic backgrounds such as Asians. Acne is a common finding, and in one study of 119 women with acne but without menstrual disorders, obesity, or hirsutism, PCOS was found on ultrasound in 45%. These findings suggest that PCOS is common in women with acne even in the absence of other suggestive clinical findings.
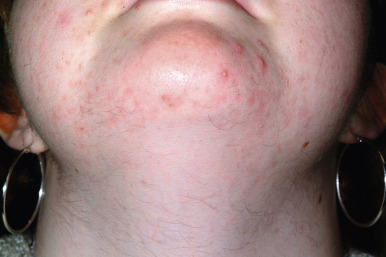
Acanthosis nigricans (AN) (see below in Acanthosis Nigricans section) is another common cutaneous finding in patients with PCOS. It presents with hyperpigmented, velvety plaques of the neck folds, axillae, and other intertriginous areas. AN is a marker for insulin resistance, although the latter appears to be only one factor leading to its development. Other important components of PCOS include menstrual irregularity and obesity. Anovulation or oligoovulation commonly occur, although women with PCOS can ovulate spontaneously. Secondary amenorrhea, oligomenorrhea, and dysfunctional uterine bleeding may all occur, and delayed menarche may be seen in girls. Adolescent females with oligomenorrhea secondary to PCOS may be difficult to distinguish from those with physiologic anovulation. Obesity is often present, with a centripetal weight distribution and increased waist-to-hip ratio of greater than 0.85. Some patients with PCOS display a metabolic pattern of atherogenic lipid profile, glucose intolerance, and increased fasting insulin level, with an increased incidence of type 2 diabetes mellitus and cardiovascular disease. Studies of adult women with type 2 diabetes mellitus show a prevalence of PCOS higher than that reported in the general population.
The clinical presentation of PCOS and its associated features have overlap with other entities described in the literature. The association of hyperandrogenism, insulin resistance, and AN has been called HAIR-AN syndrome . Although the potential causes of hyperandrogenism in this syndrome are multiple, PCOS may be present in many patients. It is possible that the etiology of insulin resistance (and resultant hyperinsulinemia) could produce both polycystic ovaries and hyperandrogenism. Syndrome X is a term that has been used to describe a systemic disease notable for hyperinsulinemia and hyperandrogenism. More recently, metabolic syndrome has been the terminology used to describe patients with abdominal obesity, dyslipidemia, glucose intolerance, and often, PCOS. These patients are also prone to hypertension, hyperuricemia, fatty liver disease, chronic inflammation, endothelial dysfunction, and coagulopathy. Importantly, there is a threefold increase in the risk of coronary heart disease and stroke. Sleep disorders such as obstructive sleep apnea and excessive daytime sleepiness have been increasingly recognized in patients with PCOS.
The diagnosis of PCOS is suggested by the clinical features of hyperandrogenism and prolonged menstrual irregularity and confirmed by finding elevated serum androgens and excluding other potential causes. The ultrasound finding of polycystic changes is common, but such changes may also be seen in normally menstruating adolescents or in other conditions, and so this must be kept in appropriate context during the evaluation. In addition, some adolescents with PCOS may not have polycystic ovaries. Screening for glucose intolerance and diabetes mellitus should be performed in all patients with PCOS, as should fasting lipid levels. Box 23-6 lists the recommended diagnostic testing for adolescents suspected of having PCOS.
Laboratory
Human chorionic gonadotropin (HCG) (urine or serum)
Follicle-stimulating hormone (FSH)
Luteinizing hormone (LH)
Estradiol
Thyroid-stimulating hormone (TSH)
Prolactin
Testosterone (total and free)
Dehydroepiandrosterone sulfate (DHEA-S)
Androstenedione
17-hydroxyprogesterone
Radiologic
Pelvic ultrasonography (transabdominal if virginal, or transvaginal)
If PCOS Confirmed:
Fasting glucose and insulin
2-hour oral glucose tolerance test
Fasting lipid panel
PCOS, Polycystic ovary syndrome.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


