Several hereditary disorders of the skin primarily manifest as disorders of the dermis. Clinical manifestations range from laxity of skin to infiltrated papules and from rigidity to thinning of dermis. Disorders of mucopolysaccharides are reviewed in Chapter 24 .
Ehlers–Danlos Syndrome
Ehlers–Danlos syndrome (EDS) consists of a group of six inherited disorders of collagen characterized by increased cutaneous elasticity, hyperextensibility of the joints, and fragility of the skin, sometimes with the formation of pseudotumors and large gaping scars. The 1997 Villefranche classification is still used with some refinements and includes autosomal dominant (AD) and autosomal recessive (AR) forms; the genetic and clinical characteristics of the major subtypes of EDS are reviewed in Table 6-1 . Most forms of EDS are linked to mutations in genes encoding fibrillar collagens, enzymes involved in posttranslational modification of these proteins, and extracellular matrix proteins. Mutations in type V and type III collagen cause classic or vascular EDS, respectively, whereas mutations involving the processing of type I collagen are involved in the kyphoscoliosis, arthrochalasis, and dermatosparaxis type of EDS.
| Type | Mutations | Skin Findings | Joint Changes | Inheritance | Other Comments |
|---|---|---|---|---|---|
| Classic * | COL5A1, COL5A2 (usually haploinsufficiency) | Hyperextensibility, bruising, velvety skin, widened atrophic scars, molluscoid pseudotumors, spheroids | Hypermobility and its complications, joint dislocations | AD | Mitral valve prolapse Hernias |
| CLASSIC VARIANTS | |||||
| EDS/OI overlap | COL1A1 | Classic EDS features | AD | Blue sclerae, short stature, osteopenia/fractures May have late arterial rupture | |
| Cardiac valvular | COL1A2 | Classic EDS features | AR | Severe cardiac valve issues as adult | |
| Periodontal | Unknown | Can have classic EDS features | Can have hypermobility | AD | Periodontitis Marfanoid habitus Prominent eyes Short philtrum |
| Progeroid | β4GALT7 or β3GalT6, encoding galactosyltransferase I or II, key enzymes in GAG synthesis | Classic EDS features | Hypermobility | AR | Loose facial skin, curly hair, alopecia, developmental delay, spondyloepimetaphyseal dysplasia with bone fragility, and severe kyphoscoliosis |
| Classic-like | TNXB | Hyperextensibility, marked hypermobility, severe bruising, velvety skin, no scarring tendency | Hypermobility | AR | Parents (especially mothers) with one TNXB mutation can have joint hypermobility |
| Hypermobility * | Unknown; some may have COL5A1 mutations | Mild hyperextensibility, scarring, textural change | Hypermobility, chronic joint pain, recurrent dislocations | AD | Sometimes confused with joint hypermobility syndrome |
| Vascular * | COL3A1 | Thin, translucent skin; bruising; early varicosities; acrogeria | Small joint hypermobility | AD | Abnormal type III collagen secretion; rupture of bowel, uterus, arteries; typical facies; pneumothorax |
| Kyphoscoliosis * | PLOD (deficient lysyl hydroxylase) | Soft, hyperextensible, bruising, atrophic scars | Hypermobility | AR | Severe muscle hypotonia, congenital kyphoscoliosis, scleral fragility and rupture, marfanoid habitus, osteopenia |
| VARIANTS WITH KYPHOSCOLIOSIS | |||||
| Spondylocheirodysplastic form | SLC39A13 , which encodes the ZIP13 zinc transporter | Similar to kyphoscoliotic form | AR | Bone abnormalities but without congenital hypotonia and progressive kyphoscoliosis Moderate short stature, wrinkled palms with thenar and hypothenar atrophy, blue sclerae | |
| Brittle cornea syndrome | ZNF469 or PRDM5 | Skin hypermobility | Joint hypermobility | AR | Kyphoscoliosis Characteristic thin, brittle cornea, ocular fragility, blue sclera, and keratoconus |
| FKBP14 deficient | FKBP14 | Features of kyphoscoliotic EDS | AR | Sensorineural hearing loss Severe congenital generalized hypotonia improved by childhood | |
| Musculocontractural | CHST14 (encoding dermatan 4-O-sulfotransferase) or DSE (encoding dermatan sulfate epimerase) | Fragile, hyperextensible skin with atrophic scars and delayed wound healing | Hypermobility | AR | Progressive kyphoscoliosis, adducted thumbs in infancy, clubfoot, arachnodactyly, contractures, characteristic facial features, hemorrhagic diathesis |
| Arthrochalasis * | Exon 6 deletion of COL1A1 or COL1A2 | Hyperextensible, soft skin with or without abnormal scarring | Marked hypermobility with recurrent subluxations | AD | Congenital hip dislocation Arthrochalasis multiplex congenita Short stature |
| Dermatosparaxis * | Type I collagen N-peptidase ADAMTS-2 | Severe fragility Sagging, redundant skin | AR | Also occurs in cattle | |
The prevalence of EDS, including the mild forms, may be as high as 1 : 5000 individuals. The spectrum of severity of EDS ranges from almost imperceptible findings to severe, debilitating disease. Most common is the classical type ( Box 6-1 ). The vascular type tends to be most devastating. The kyphoscoliosis, arthrochalasis, and dermatosparaxis types are considerably less common than other forms. The effect on quality of life can be considerable, and three-quarters of affected individuals report fatigue that may be impacted by pain, sleep disturbance, difficulty with concentration, and diminished social functioning.
- ▪
Autosomal dominant
- ▪
Skin
- ▪
Hyperextensible
- ▪
Velvety, doughy skin
- ▪
Fragile, thin, poor tensile strength, gaping scars after wounding
- ▪
Easy bruisability
- ▪
Pseudotumors and spheroids
- ▪
Piezogenic papules
- ▪
- ▪
Joints/extremities
- ▪
Hypermobility
- ▪
Sprains, dislocations
- ▪
Pes planus
- ▪
Chronic joint and limb pain
- ▪
Muscle hypotonia, delayed motor development
- ▪
- ▪
Hiatal hernia, anal prolapse
- ▪
Mitral valve prolapse
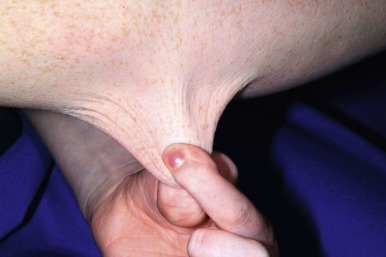
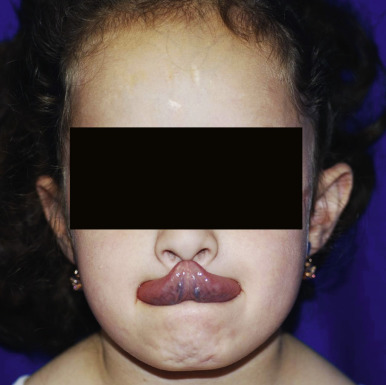
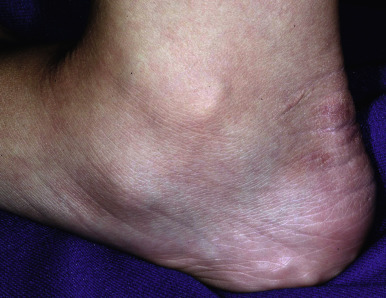
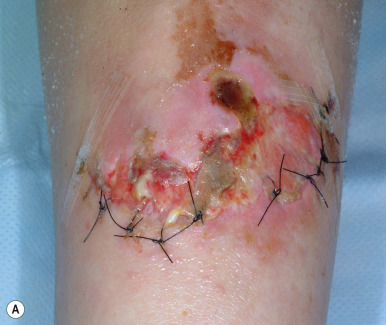
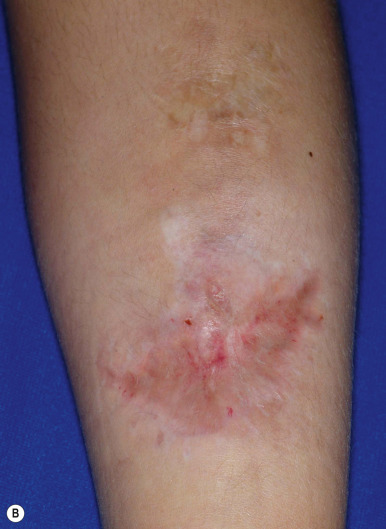
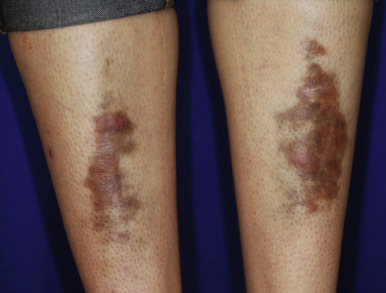
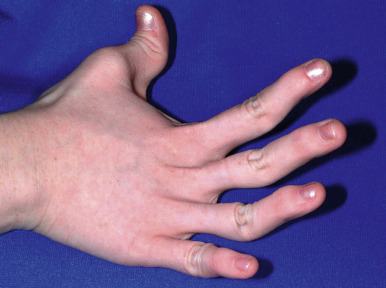
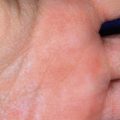
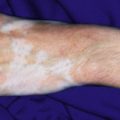
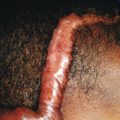
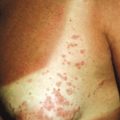
The skin of individuals with the classic type is velvety, soft, and has a doughy consistency. After being stretched, it returns to its normal position as soon as released, in contrast to the lax skin of cutis laxa. Skin hyperextensibility should be tested at a site not subjected to mechanical forces or scarring, such as the volar surface of the forearm ( Fig. 6-1 ). In contrast to 10% of apparently normal individuals, approximately 50% of patients with EDS can touch the tip of their nose with their tongue (Gorlin sign) ( Fig. 6-2 ). In addition, the skin of the hands, feet, and at times the elbows tends to be lax and redundant, thus resulting in a loose-fitting glove- or moccasin-like appearance. A number of patients have pressure-induced herniation of subcutaneous fat on the wrists or on the medial or lateral aspect of the heels, evident when the patient is standing (piezogenic pedal papules) ( Fig. 6-3 ). In addition to abnormal elasticity, the skin of patients with the classical (but not the hypermobile) form of EDS is extremely fragile, and minor trauma may produce gaping “fishmouth” wounds ( Fig. 6-4 ). It has poor tensile strength and cannot hold sutures properly. This leads to dehiscence, poor healing, and the formation of wide, papyraceous, wrinkled hernia-like scars, particularly over areas of trauma (such as the forehead, elbows, knees, and shins). Blood vessels are fragile, resulting in hematomas and a chronic bruise-like appearance, particularly on the anterior aspect of the lower extremities ( Fig. 6-5 ). The resolution of hematomas is accompanied by fibrosis, which produces soft subcutaneous nodules (pseudotumors) and calcified subcutaneous nodules, especially on the shins and forearms (spheroids). Hyperextensible joints may result in “double-jointed” fingers or subluxation of larger joints ( Fig. 6-6 ). This may occur spontaneously or follow slight trauma. Generalized hypermobility is determined by a score of 5 or higher in the 9-point scale established by Beighton and colleagues ( Fig. 6-7 ; Table 6-2 ). Sprains, dislocations or subluxations, and pes planus occur as complications, and patients complain of chronic joint and limb pain despite normal skeletal radiographs. Muscle hypotonia and delayed gross motor development have been described. Hiatal hernia, postoperative hernias, and anal prolapse have been noted as manifestations of the tissue hyperextensibility and fragility. Anomalies of the heart and dissecting aortic aneurysms have rarely been described in the classical form in contrast to the arterial form. Mitral valve prolapse is a common manifestation, but aortic root dilation is uncommon; both can be assessed by echocardiography, computed tomography (CT), or magnetic resonance imaging (MRI) examinations.

| Finding | Negative | One Side | Both Sides |
|---|---|---|---|
| Passive flexion of thumb to forearm | 0 | 1 | 2 |
| Passive dorsiflexion of fifth finger >90 degrees | 0 | 1 | 2 |
| Hyperextension of elbows >10 degrees | 0 | 1 | 2 |
| Hyperextension of knees † >10 degrees | 0 | 1 | 2 |
† Touching the floor with palms open and knees fully extended.
The diagnosis of classical EDS is primarily clinical. Routine histopathologic examination of skin from patients with EDS is usually normal but may show loose collagen and irregular fibroblasts ; electron microscopy demonstrates disorganized collagen fibers and fibrils with variable cross-sectional diameters. Studies of platelet function and coagulation are usually normal despite a tendency for bruising and increased bleeding, further implicating the defective structural integrity of skin and blood vessels.
The underlying molecular defect for 93% of patients with the classical form of EDS is mutations in the α1 or α2 chain of type V collagen, often leading to haploinsufficiency. Type V collagen is a minor fibrillar collagen that regulates collagen fibril diameter. Patients with mutations in COL1A1 at a nonglycine site (mutations at glycine sites lead to osteogenesis imperfecta [OI]) may exhibit features of classic EDS alone or with subtle features of OI (blue sclera, relatively short stature, osteopenia, or fractures). Some children with nonglycine substitutions in type I collagen have classic EDS but develop spontaneous arterial rupture during adulthood.
Several variants resemble classical EDS (see Table 6-1 ). A “classiclike” autosomal recessive form of EDS, caused by deficiency of the extracellular matrix protein tenascin-X, shows typical EDS but normal wound healing. Heterozygous carriers may exhibit only joint hypermobility. A contiguous gene syndrome caused by a 6p deletion can present with tenascin-X deficiency as well as congenital adrenal hyperplasia (deletion in CYP21A2 ) with or without ovarian failure (deletion in MSH5 ). The periodontal type (formerly type VIII EDS) is a variant of the classic type of EDS and shows variable inter- and intrafamilial clinical manifestations (including patients without skin or joint abnormalities). The periodontal type is associated with periodontitis, a marfanoid body habitus, prominent eyes, and a short philtrum. Patients with progeroid EDS (PEDS) exhibit loose, wrinkled facial skin; fine, curly hair; sparse eyebrows and eyelashes; downslanting palpebral fissures; developmental delay; skeletal abnormalities (spondyloepimetaphyseal dysplasia with bone fragility and severe kyphoscoliosis); and cutaneous and joint findings of classical EDS. This variant is caused by biallelic mutations in either galactosyltransferase I (β4GALT7) or II (β3GalT6), key enzymes in initiating glycosaminoglycan (GAG) synthesis.
Patients with the hypermobility type show extensive joint hypermobility and dislocations, particularly of the shoulder, patella, and temporomandibular joints. The skin is often soft and velvety but heals well and only occasionally shows hyperextensibility. Given that up to 40% of school-age children may show a score of 5 or higher using the 9-point Beighton criteria, the diagnosis of hypermobility-type EDS may be difficult. It is also hard to distinguish from benign joint hypermobility syndrome, and the term joint hypermobility syndrome has been suggested to encompass any child with symptomatic joint hypermobility, including those with hypermobility-type EDS. The autonomic burden of hypermobility-type EDS is as high as in fibromyalgia and is primarily characterized by orthostatic issues (dizziness or even transient loss of consciousness with postural change or standing), palpitations, shortness of breath, and gastrointestinal (GI) complaints.
The vascular type of EDS is characterized by extensive bruising and often thin, translucent skin that is not hyperextensible. Joint hypermobility is usually limited to the digits, and only 17% of patients meet Beighton criteria. The facial appearance is often typical, with sunken eyes, a thin upper lip, and decreased facial fat. Spontaneous rupture of arteries, particularly midsized arteries, may occur during childhood, although its peak age of incidence is the third or fourth decade of life. Arterial or intestinal rupture often presents as acute abdominal or flank pain (including from GI bleeding or bowel perforation), and intracranial aneurysms may be associated with cerebrovascular accidents. Arterial rupture is the most common cause of death. Pregnancies may be complicated by prepartum and postpartum arterial bleeding and by intrapartum uterine rupture. Vaginal and perineal tears from the delivery heal poorly, and pneumothorax occurs in 11% of affected individuals. In one series, 94% of patients with vascular EDS showed blue sclerae.
The vascular form of EDS has been associated with mutations in type III collagen ( COL3A1 ), resulting in a reduced amount of type III collagen in dermis, vessels, and viscera. The prognosis for individuals with null mutations in COL3A1 is better than with missense or exon-skipping mutations.
Vascular EDS may be confused with periodontal EDS and Loeys–Dietz syndrome. The latter is an autosomal dominant disorder characterized by joint hypermobility with dislocations; soft velvety skin; arterial tortuosity; widespread vascular aneurysm and dissection ; and an increased risk of atopy. The disorder has been divided into Loeys–Dietz syndrome type 1 and type 2. Patients with type 1 disease have a marfanoid habitus (but do not fulfill Ghent criteria for Marfan syndrome [MFS]), craniofacial abnormalities, and mutations in TGFBR1 (transforming growth factor [TGF]-β1 receptor), whereas patients with type 2 have mutations in TGFB2R and no craniofacial or skeletal anomalies.
The kyphoscoliotic type of EDS is characterized by generalized joint laxity with severe muscle hypotonia at birth, which leads to gross motor delay and congenital progressive scoliosis. Severe hypotonia is predominant during infancy, and patients are thought to have congenital muscular dystrophy or myopathies. By the second or third decade of life, patients tend to lose the ability to ambulate. The skin may be fragile and heals with atrophic scars. Easy bruisability and arterial rupture have been described. Patients often show a marfanoid habitus, osteopenia, and scleral fragility with rupture of the ocular globe. Because of PLOD1 mutations, this form shows a deficiency of collagen lysyl hydroxylase. The diagnosis can be confirmed by finding a high ratio of lysyl pyridinoline (LP) to hydroxylysyl pyridinoline (HP) crosslinks (LP/HP) in the urine by high-performance liquid chromatography (HPLC).
The spondylocheirodysplastic form of EDS resembles the kyphoscoliotic form but lacks the severe muscular hypotonia from birth and progressive kyphoscoliosis. In contrast to the kyphoscoliotic form, the spondylocheirodysplastic form shows moderate short stature, wrinkled palms with thenar and hypothenar atrophy, blue sclerae but no other eye abnormalities, and a variety of bone abnormalities. Biallelic mutations in SLC39A13, which encodes the zinc transporter protein ZIP13, important for intracellular zinc homeostasis, are causative.
Musculocontractural EDS is a variant with characteristic facial features, multiple contractures, a hemorrhagic diathesis with large subcutaneous hematomas, and several other systemic complications; this variant has been linked to dysfunction of dermatan sulfate from mutations in both alleles of genes encoding dermatan 4-O-sulfotransferase (CHST14) or dermatan sulfate epimerase (DSE). Another variant with progressive kyphoscoliosis, congenital hypotonia, joint hypermobility, and hyperelastic skin results from mutations in FK506 -binding peptidylprolyl cis-transisomerases ( FKBP14 ) ; the presence of sensorineural hearing loss aids in diagnosis. The brittle cornea syndrome (BCS), an autosomal recessive condition caused by mutations in either ZNF469 or PRDM5, is characterized by thin, brittle cornea; ocular fragility; blue sclera; and keratoconus; as well as associated skin and joint hypermobility and kyphoscoliosis. Lysyl hydroxylase production and urinary pyridinoline excretion are normal in these variants.
Patients with the arthrochalasis type show generalized joint hypermobility with severe recurrent subluxations and congenital bilateral hip dislocation. The skin may be hyperextensible, fragile, and easy to bruise. Early and significant muscle hypotonia, kyphoscoliosis, and mild osteopenia have been described. Mutations in COLA1A1 or COL1A2 that result in skipping of exon 6 lead to defective collagen synthesis.
The dermatosparaxis form of EDS is characterized by severe skin fragility with sagging, redundant skin ; wound healing leads to wide atrophic scars, especially past infancy. The skin may be soft and doughy in texture with easy bruisability. Characteristic facies, gingival hyperplasia, large umbilical and inguinal hernias, delayed closure of the fontanels, postnatal growth failure, and premature rupture of the fetal membranes may be seen. Electrophoretic demonstration of procollagen α1(I) or α2(I) chains from collagen or cultured fibroblasts is seen with the arthrochalasis and dermatosparaxis forms.
Management of Ehlers–Danlos Syndrome
Management is mainly supportive, but cardiovascular work-up, physiotherapy, pain management, and psychological and genetic counseling may be important. Wearing a medical bracelet warning of EDS and carrying information for teachers and healthcare workers can be helpful. The Ehlers–Danlos National Foundation ( www.ednf.org ) is a national support group. Children with skin fragility should use protective pads on the shins, knees, and forehead to decrease the risk of laceration and bruising. Low-impact sports (such as swimming) are preferable to contact sports and weight training in patients with hypermobility. Physical and occupational therapists or rheumatologists can suggest devices (such as splints and braces) to make movement more comfortable. Children with significant hypotonia or motor developmental delay should be referred for appropriate physical therapy to improve muscular strength and coordination. Pain medication should be tailored to individual needs. Psychological counseling to help develop coping strategies and treat depression can be important, especially in patients with hypermobility and chronic pain.
A baseline echocardiogram should be performed in all patients with EDS in order to evaluate aortic root diameter and cardiac valves. Echocardiograms should be repeated in children and adolescents approximately every 3 years if the results are normal and annually if results are abnormal. Patients who have mitral valve prolapse and regurgitation should be given prophylactic antibiotics to prevent bacterial endocarditis.
Surgical procedures present problems, because tissues are friable and difficult to suture. Therefore edges of wounds should be approximated without tension by closely spaced sutures in two layers (absorbable and retained), adhesive reinforcement (e.g., Steri-Strips) to minimize scar spread, and pressure bandages to aid healing, diminish scarring, and lower the risk of hematoma and pseudotumor formation. Sutures should be kept in place for at least twice as long as for normal skin, and use of absorbable suture material without removal has been proposed. Prophylactic antibiotics after injury and close monitoring for postoperative infection are important to minimize postoperative complications. Ascorbate is a cofactor that stimulates prolyl hydroxylase and lysyl hydroxylase, enzymes that catalyze the formation of hydroxyproline and hydroxylysine to promote collagen deposition. Doses of 2 to 4 g/day have been administered to patients with the kyphoscoliosis type to improve wound healing and decrease the bleeding tendency. Ascorbic acid sometimes decreases the bleeding tendency in other types. Anti-inflammatory drugs may improve the musculoskeletal pain associated with EDS, but those interfering with platelet function and prolonging bleeding (acetylsalicylic acids, nonsteroidal anti-inflammatory drugs [NSAIDs] other than celecoxib family) should be avoided in patients with significant bruising. Dental procedures require extra precautions as well, because of increased bleeding. More than half of patients who undergo posterior spinal fusion for scoliosis develop various complications.
In patients with the vascular type of EDS, invasive vascular procedures (such as arteriography and catheterization) increase the risk of vascular rupture and should be replaced by ultrasound and/or subtraction angiography. Surgery should be avoided if possible but may be required for arterial or bowel complications. Manipulation of vascular and other tissues should be minimized if surgery is required. Celiprolol, a long-acting β 1 antagonist and partial β 2 agonist, decreased by threefold the incidence of arterial rupture or dissection in patients with vascular EDS. Pregnancy for women with the vascular type of EDS is high risk (up to 12% die from uterine rupture or peripartum arterial rupture). Planned caesarean section may be preferable to vaginal delivery. Infants with EDS are prone to premature birth because of early rupture of membranes. Preterm delivery occurs in 40% of pregnancies with affected neonates and 21% of pregnancies of affected mothers.
Marfan Syndrome
MFS is an AD disorder that occurs in approximately 1 : 5000 persons and affects primarily the skeletal, ocular, and cardiovascular systems. Mutations primarily occur in the FBN1 gene that encodes fibrillin-1 (66% to 91%) and rarely in TGFBR1 or TGFBR2, the genes also associated with Loeys–Dietz syndrome (see Ehlers–Danlos Syndrome section). Fibrillin-1 is secreted into the extracellular matrix and polymerizes to form microfibrils of the zonular fibers of the lens and are associated with elastic fibers in the aorta and skin. The microfibrils stabilize latent TGF-β-binding proteins (LTBPs), which bind to and maintain TGF-β in an inactive state. Deficiency of fibrillin-1 thus leads to TGF-β activation. About 27% of cases occur by spontaneous mutation; recurrence as a result of parental germline mosaicism has been described.
Diagnosis is best made by clinical examination (including careful cardiac evaluation and measurement of body proportions) ( Box 6-2 ), echocardiography, slit-lamp ophthalmologic examination, and radiographs or imaging as needed to find criteria. Cardiac examinations should be repeated every year until adulthood. Early diagnosis is critical because of the potentially fatal complications, but the clinical features evolve with advancing age and diagnosis is difficult. Family history is also important, but manifestations may not be evident until adolescence and expressivity is variable, so that generations appear to be skipped. The Ghent diagnostic criteria ( Box 6-3 ) were simplified in 2010, eliminating major and minor criteria and giving more weight to ectopia lentis and aortic root aneurysm/dissection. Other organ involvement contributes to a systemic score to guide diagnosis if aortic disease, but not ectopia lentis, is present. A more important role is assigned to genotyping FBN1 and other relevant genes. Criteria now enable diagnosis of ectopia lentis; mitral valve prolapse and myopia, mild and nonprogressive aortic root dilation, marfanoid skeletal changes, and skin features (MASS); and mitral valve prolapse syndrome. Criteria are still less reliable in children, especially since several of the more specific features are age-dependent (e.g., aortic dilation, ectopia lentis, dural ectasia, protrusion acetabuli), and care should still be made to avoid branding children unnecessarily with MFS, given the potential stigma and lifestyle restrictions.
- ▪
Autosomal dominant disorder
- ▪
Present in ≥75% of individuals with Marfan syndrome (MFS):
- ▪
Positive wrist and thumb sign
- ▪
- ▪
Present in ≥50% and <75% of individuals with MFS:
- ▪
Dolichostenomelia *
* Dolichostenomelia refers to a habitus in which the limbs are unusually long, as is typical of MFS ( dolichos, long; steno, narrow or close; melia, of the limbs).
- ▪
Pes planus
- ▪
Joint hypermobility
- ▪
Aortic root dilation
- ▪
Mitral valve prolapse
- ▪
High arched palate with crowded dentition
- ▪
Typical facial features
- ▪
Lens displacement (ectopia lentis)
- ▪
- ▪
Present in ≥25% and <50% of individuals with MFS:
- ▪
Pectus excavatum or carinatum requiring surgery
- ▪
Myopia
- ▪
Scoliosis
- ▪
Striae
- ▪
Without a family history of Marfan syndrome (MFS), any of these combinations allow diagnosis:
- 1.
Aortic diameter (Z score *
* Z score is the standard deviation from normal means of the inner to inner-edge diameter of the aortic root at the sinus of Valsalva, normalized for the subject’s body surface area and age.
≥2) AND ectopia lentis
- 2.
Aortic diameter (Z ≥ 2) AND fibrillin-1 ( FBN1 ) mutation
- 3.
Aortic diameter (Z ≥ 2) AND systemic criteria (≥7 points; see criteria below)
- 4.
Ectopia lentis AND fibrillin-1 ( FBN1 ) mutation with known aortic diameter (Z ≥ 2)
With a positive family history, these features allow the diagnosis:
- 1.
Ectopia lentis
- 2.
Systemic criteria (≥7 points) (see criteria below)
- 3.
Aortic diameter (Z ≥ 2 above 20 years old, ≥3 below 20 years old)
Alternative diagnoses (no family history of MFS):
Ectopia lentis syndrome: ectopia lentis with or without systemic criteria AND: (1) with an FBN1 mutation but one not known to be associated with aortic diameter (Z ≥ 2) OR (2) no FBN1 mutation
MASS: aortic diameter (Z < 2) AND systemic criteria (≥5 with at least one skeletal feature) but without ectopia lentis
MVP syndrome: MVP AND aortic diameter (Z < 2) AND systemic criteria (<5) without ectopia lentis
Systemic criteria (maximum total: 20 points; score >7 indicates systemic involvement):
Wrist AND thumb sign = 3 (wrist OR thumb sign = 1)
Pectus carinatum deformity = 2 (pectus excavatum or chest asymmetry = 1)
Hindfoot deformity = 2 (plain pes planus = 1)
Pneumothorax = 2
Dural ectasia = 2
Protrusio acetabuli = 2
Reduced US/LS AND increased arm/height AND no severe scoliosis = 1
Scoliosis or thoracolumbar kyphosis = 1
Reduced elbow extension = 1
Facial features (3/5) = 1 (dolichocephaly, enophthalmos, downslanting palpebral fissures, malar hypoplasia, retrognathia)
Skin striae = 1
Myopia >3 diopters = 1
MVP (all types) = 1
MASS, Mitral valve prolapsed and myopia, mild and nonprogressive aortic root dilation, marfanoid skeletal changes, and skin features; MVP, mitral valve prolapse; US/LS, upper segment/lower segment ratio.
Patients are often tall with long extremities (“marfanoid habitus”; see Fig. 6-7 ). The arm span characteristically is greater than the height, and after puberty the upper segment (vertex to pubis)-to-lower segment (pubis to sole) ratio is less than 0.86. Arachnodactyly, kyphoscoliosis, pectus carinatum or excavatum, and a hindfoot deformity or flat feet are commonly seen in patients with this disorder. Joint laxity from capsular, ligamentous, and tendinous involvement may cause hyperextensibility and/or dislocation. Patellar dislocation is not uncommon; dislocation of the hip, often detected during the newborn period, may be the first sign of MFS. The thumb sign (thumb extends well beyond the ulnar border of the hand when overlapped by fingers) and wrist sign (thumb overlaps the fifth finger as they grasp the opposite wrist) are screening tests for the joint hypermobility of MFS. Dural ectasia is occasionally seen in children, and imaging is performed largely to consider an additional Ghent criterion for diagnosis, but its presence is nonspecific. Specific facial features and pneumothorax are additional criteria (see Box 6-3 ).
Lack of subcutaneous fat and the presence of striae, most prominent on the upper chest, arms, thighs, and abdomen, are the most common cutaneous manifestations of MFS. In addition, elastosis perforans serpiginosa (see Elastosis Perforans Serpiginosa section ) occurs with increased incidence in patients with MFS.
The most common ocular abnormalities are lens displacement (ectopia lentis; the hallmark of ocular involvement, seen in at least 55% of affected children) and myopia (almost half of children). Retinal detachment, cataracts, or glaucoma may impair vision and cause blindness. Cardiovascular abnormalities occur in almost 70% of children with MFS. Dilation of the aorta is the most common defect and is generally greatest at the sinuses of Valsalva, and diffuse dilation of the proximal segment of the ascending aorta with aortic regurgitation often occurs. Mitral valve prolapse occurs in approximately 60% of affected children and adolescents. Left ventricular dilation may predispose to patients to alterations of repolarization and fatal ventricular arrhythmias. The most severely affected children, those with neonatal MFS, almost always have aortic dilation and severely impaired valves, leading to congestive heart failure, pulmonary hypertension, and a risk of early death. In a child diagnosed with MFS, serial echocardiography at 6- to 12-month intervals is recommended, the frequency depending on the aortic diameter in relation to the body surface area and the rate of increase. Pregnancy has been associated with significant cardiac complications (aortic dissection, arrhythmia), postpartum hemorrhage, thromboembolism, and premature and babies who are small for gestational age.
Conditions most often considered when patients have cutaneous and other system concerns are vascular-type EDS (aortic root dilation or dissection), Loeys–Dietz syndrome (aortic root dilation or dissection), homocystinuria, and MASS. Patients with MFS do not have the translucent and velvety skin or easy bruising of EDS or Loeys–Dietz syndrome. Neither type I nor type II Loeys–Dietz syndrome includes myopia or ectopia lentis, and craniosynostosis, hypertelorism, and bifid uvula, features of Loeys–Dietz syndrome, are not features of MFS. Homocystinuria is an autosomal recessive disorder caused by mutations cystathionine β-synthase in which patients also show ectopia lentis and a marfanoid habitus; the presence of mental retardation in patients with homocystinuria is a distinguishing feature. A defect in cobalamin C, an essential cofactor for cystathionine β-synthase, similarly presents with marfanoid features, arachnodactyly, joint laxity, and scoliosis, in addition to macrocytic anemia and developmental delay.
Perhaps hardest to distinguish is MASS, a marfan-like disorder caused by FBN1 mutations but with a better prognosis. Striae are a prominent feature, and atrophic skin patches with abnormal elastic fibers may be present. Pectus excavatum and marfanoid skeletal features are most common. Among the distinguishing features are a stable and mild aortic root dilation and lack of ectopia lentis.
The prognosis of MFS depends on the extent and severity of cardiovascular defects. Death usually occurs in adulthood, but occasionally during childhood, as a result of cardiovascular sequelae, especially owing to complications related to dilation of the aortic root. A rare neonatal form of MFS features the marfanoid body disproportion, lax skin, emphysema, ocular abnormalities, joint contractures, kyphoscoliosis, adducted thumbs, crumpled ears, micrognathia, muscle hypoplasia, and deficient subcutaneous fat over joints. Severe cardiac valve insufficiency and aortic dilation result in death during the first 2 years of life. A neonatal progeroid variant of MFS with prematurity, congenital lipodystrophy, and frameshift mutations at the 3′ end of FBN1 has been described. In the differential diagnosis of progeria, the marfanoid habitus with accelerated linear growth, severe myopia, and dilation of the aortic bulb allow correct diagnosis.
Children with MFS but without serious cardiac issues can participation in some sports but should avoid potentially harmful exertion, particularly contact sports (to protect the aorta and lens), sports with bursts of activity (such as sprinting), and isometric exercises (such as weightlifting), which might lead to further aortic root dilation, aortic rupture, or congestive heart failure. Scuba diving should be avoided because of the risk of pneumothorax. Long-term propranolol therapy may decrease myocardial contractility, thus decreasing the risk of aortic dilation, but the relative value of β-blockers versus the angiotensin receptor blocker losartan (or the combination of the two) is undergoing testing. In one study, long-term treatment with doxycycline was shown to be more effective than a β-blocker in preventing thoracic aortic aneurysm through its inhibition of matrix metalloproteinases-2 and -9. Aneurysmal and valvular heart defects may require prosthetic replacement, but this should be postponed as long as possible to avoid recurrent prosthesis replacement, particularly in growing children. Prophylactic replacement of the aortic root to prevent aortic dissection has led to increased life expectancy; in one recent study striae together with elevated TGF-β and matrix metalloproteinase (MMP)-3 serum levels correlated with a higher risk of aortic dissection. Early and regular ophthalmologic examinations are required to detect correctable amblyopia and retinal detachment. Ectopia lentis and even complete luxation may be tolerated for decades, but lens extraction may be required to treat diplopia, glaucoma, cataracts, or retinal detachment. Repair of the pectus excavatum is indicated if cardiopulmonary compromise occurs but should be delayed until skeletal maturation is nearly complete to prevent recurrence and should employ internal stabilization. Scoliosis may be lessened in adolescent girls by estrogen therapy, but this therapy may also produce an overall decrease in height. Bracing, physical therapy, and vertebral fusion may all be necessary to prevent severe scoliosis. The website for the National Marfan Foundation is www.marfan.org .
Osteogenesis Imperfecta
OI refers to a group of inherited disorders of bone fragility in which the skin may be thin, atrophic, and somewhat translucent. The easy bruisability and bone fractures may raise the possibility of child abuse. Wound healing may be normal, but scars are commonly atrophic or hypertrophic. Patients often show hyperlaxity of ligaments and hypermobility of joints, but joint dislocation does not occur. Blue sclerae are distinctive features in approximately 90% of patients but can also be seen in MFS and EDS; increased electron-dense granular material between scleral fibers permits scattering of light by normal pigment within the orbit. Otosclerosis with hearing loss may begin during adolescence (50% of the common type I by adulthood), and fragile, discolored teeth are particularly common in the more unusual forms (dentinogenesis imperfecta [DI]). Patients may show mitral and aortic valve dilation and regurgitation and cystic medionecrosis of the aorta.
More than one classification scheme has recently been proposed; the newer Sillence classification includes types 1 through 5 and the more-newly recognized, less common, and largely autosomal recessive forms based on clinical features. The classification described by Marini et al. separates types of OI based on genetics and extends the five Sillence classification subtypes to 12 subtypes (I through XII) and an unclassified/other category ( Table 6-3 ). OI has also been classified by severity of clinical features (mild to extremely severe), fracture frequency, bone densitometry, and level of mobility. Changes in 16 genes may be classified under OI, presenting variable clinical features, severity, and prognosis. More than 85% of affected individuals have AD OI, of which most are type I OI, which is by far the mildest form.
| Type | Frequency/Inheritance | Gene; Protein | Clinical Features | Effect of Gene Alteration |
|---|---|---|---|---|
| I | AD (>50%) | COL1AI, COL1A2 ; type I collagen | Also called nondeforming OI with blue sclerae Fractures (90% to 95%), blue sclerae (100%), hearing loss (begins in adolescence and is in >50% by 40 years), vertigo, normal stature, rarely DI | Collagen I quantity |
| II | AD * | COL1AI, COL1A2I glycine substitutions; type I collagen | Also called perinatal lethal OI Multiple fractures, severe rib (beading) and long bone (crumped, accordion-like) deformities, frog leg positioning, blue-gray sclerae, small for gestational age 20% stillborn and 40% die by 1 month | Collagen I structure |
| III | AD | COL1AI, COL1A2I glycine substitutions; type I collagen | Also called progressive deforming OI Severe deformities; multiple fractures with progressive deformities, short stature (<3rd %) with severe progressive scoliosis, marked osteopenia, nonambulatory Triangular face, hearing loss in adults, may have DI Progressive whitening of sclerae Majority now survive into adulthood and have normal growth velocity/decreased fractures with cyclic IV bisphosphonates | Collagen I structure |
| IV | AD | COL1AI, COL1A2I glycine substitutions; type I collagen | Also called common variable OI ; milder than OI III Moderately short, mild to moderate scoliosis, recurrent fractures with variable deformity, osteoporosis Typically ambulatory DI common, adult-onset hearing loss, usually normal sclerae | Collagen I structure |
| V | AD (4% to 5%) | IFITM5 ; BRIL | Also called OI with calcification in interosseous membranes Mild to moderate fractures (especially forearms, legs affected) and short stature, dislocation of radial head White sclerae, no DI Rarely, painful hyperplastic callus | Matrix mineralization |
| VI | AR | SERPINF1 ; PEDF | Similar to type III, moderately short, scoliosis, white sclerae, no DI Abnormal bone mineralization | Matrix mineralization |
| VII | AR | CRTAP ; cartilage-associated protein | Moderately deforming Clinically types II and III, but milder forms documented Rhizomelic shortening of humerus and femur, coxa vara, “popcorn epiphyses” White sclerae, no DI | 3-hydroxylation of collagen |
| VIII | AR | LEPRE1 ; prolyl-3-hydroxylase-1/P3H1 | Moderately deforming Clinically types II and III, but milder forms documented Rhizomelic shortening of humerus and femur, coxa vara, “popcorn epiphyses” White sclerae, no DI | 3-hydroxylation of collagen |
| IX | AR | PPIB ; peptidyl-prolyl isomerase B/CyPB | Clinically types II and IV Fractures with generalized osteopenia, wide anterior fontanel Moderate growth deficiency, gross motor delay No rhizomelia or DI, white sclerae | 3-hydroxylation of collagen |
| X | AR | SERPINH1 ; HSP47 | Clinically type III | Collagen chaperone |
| XI | AR | FKBP10 ; FKBP10 (also called FKBP65) | Clinically type III Can have joint contractures | Telopeptide hydroxylation for crosslinking |
| XII | AR | BMP1 ; BMP1/mTLD | Clinically type III | Collagen processing (cleaves C-propeptidase) |
| OTHERS | ||||
| AR | CREB3L1 ; OASIS | Clinically type III | Osteoblast development | |
| AR | SP7/OSX ; SP7/osterix | Clinically type II | Osteoblast development | |
| AR | TMEM38B ; TRIC-B | Clinically type III | Osteoblast development | |
| AD/AR | WNT1 ; WNT1 | Clinically type II (AD) or type III (AR) | Osteoblast development | |
| AR | PLOD2 ; PLOD2 | Clinically type III; can have congenital joint contractures | Telopeptide hydroxylation | |
| XLR | PLS3 ; plastin 3 | Clinically closest to type IV; no features beyond childhood fractures; manifests in hemizygous males and variable in heterozygote females | Bone formation and remodeling | |
* Usually new mutation; parental germline mosaicism in 2% to 6% of cases.
Patients with AD OI almost all have mutations in type I collagen, which is composed of two α1 (COLIA1) and one α2 (COLIA2) polypeptide chains, forming a triple helical structure. Dominant OI can also result from mutations in IFITM5, leading to abnormalities in matrix mineralization. Overall, 10% to 15% of cases are autosomal recessive and result from mutation in one of at least 11 genes involved in posttranslational processing of type I collagen and bone formation (see Table 6-3 ). Some of these encode proteins involved in a complex required for proline (P986) hydroxylation of type I collagen (CRTAP, P3H1, CYPB) . Others are involved in collagen mineralization ( BRIL and PEDF ), collagen crosslinking, folding and chaperoning ( FKBP65 and HSP47 ), and osteoblast development (osterix, OASIS, TRICB and WNT1 ) (see Table 6-3 ).
Supportive orthopedic therapy includes physical therapy to prevent contractures and immobility-induced bone loss, orthoses to protect the lower limbs, and in more severe cases bisphosphonates. Cyclical treatment with intravenous bisphosphonates is the mainstay of medical therapy for children with moderate to severe OI and has been shown to increase bone mass density, improve muscle strength and mobility, and decrease fractures and bone pain.
Cutis Laxa
Cutis laxa (generalized elastolysis) results from mutations in one of 11 genes that disrupt the elastic tissue network in skin ( Box 6-4 ; Table 6-4 ). In general, the products of these genes are either: (1) proteins involved in the assembly of elastic fibers and upregulation of TGF-β signaling (e.g., ELN, FBLN4, FBLN5, and LTBP4 ); (2) Golgi proteins that facilitate secretion of elastic network components (e.g., ATP6V0A2, RIN2, and GORAB ); or (3) mitochondrial proteins (e.g., PYCR1, ALDH18A1, and SLC2A10 ).
- ▪
Autosomal dominant or autosomal recessive forms
- ▪
Autosomal recessive forms subdivided into those with cardiovascular features (ARCL-1), those with developmental delay (ARCL-2), and those with developmental delay, athetosis, and corneal clouding (ARCL-3). A variety of underlying molecular bases for each group have been discovered (see Table 6-4 )
- ▪
Skin
- ▪
Loose, inelastic skin
- ▪
- ▪
Most common other features:
- ▪
Facial dysmorphism
- ▪
Aortic dilation
- ▪
Pulmonary artery stenosis
- ▪
Pulmonary emphysema
- ▪
Diverticulae: gastrointestinal, genitourinary
- ▪
Uterine or rectal prolapse
- ▪
Ventral, hiatal, inguinal hernias
- ▪
ARCL, Autosomal recessive cutis laxa.
| Disorder | ADCL | ARCL-1A | ARCL-1B | ARCL1C/URDS | ARCL-2A | ARCL-2B | ARCL-3/De Barsy | GO | XLCL | MACS | ATS |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | ELN | FBLN4 | FBLN5 | LTBP4 | ATP6V0A2 | PYCR1 | ALDH18A1; PYCR1 | GORAB | ATP7A | RIN2 | SLC2A10 |
| Protein | Elastin | Fibulin-4 | Fibulin-5 | LTBP4 | ATP6V0A2 | PYCR1 | P5CS; PYCR1 | GORAB | ATP7A | RIN2 | GLUT10 |
| Lax skin | +++ * | ++ | +++ | +++ | +++ | +++ | +++ | ++ | +++ | ++ | +++ |
| Facial features | — | ||||||||||
| Prominent ears | +++ | + | +++ | + | — | — | — | — | ++ | + | |
| Hypertelorism | — | +++ | — | +++ | — | — | — | — | — | ++ | |
| Retrognathia | — | +++ | — | +++ | +++ | +++ | — | — | — | +++ | |
| Other dysmorphism | — | ++ | — | ++ | — | ++ | ++ | +++ | ++ | ++ | |
| Emphysema | ++ | ++ | +++ | +++ | — | — | — | — | — | — | — |
| Supravalvular aortic stenosis | — | — | +++ | — | — | — | — | — | — | — | — |
| Aortic aneurysm | ++ | +++ | — | — | + | — | — | — | — | — | ++ |
| Arterial tortuosity | — | +++ | + | — | — | — | ++ | — | ++ | — | +++ |
| Hernias | ++ | +++ | +++ | +++ | +++ | ++ | ++ | ++ | +++ | + | ++ |
| Bladder diverticula | — | — | + | +++ | + | — | — | — | +++ | — | — |
| Delayed motor development | — | + | + | — | +++ | — | +++ | +++ | +++ | — | — |
| Mental retardation | — | — | — | — | +++ | +++ | +++ | ++ | +++ | — | — |
| Growth delay | — | ++ | + | +++ | +++ | +++ | +++ | +++ | + | — | — |
| IUGR | — | — | — | + | +++ | +++ | +++ | +++ | — | — | — |
| Lax joints | — | + | — | ++ | +++ | +++ | ++ | +++ | ++ | +++ | +++ |
| Hypotonia | — | + | — | +++ | +++ | — | ++ | ++ | ++ | — | — |
| Congenital hip dislocation | — | + | — | — | + | +++ | ++ | ++ | — | — | — |
| Patent anterior fontanel | — | — | — | ++ | +++ | — | ++ | ++ | ++ | — | — |
| Occipital horns | — | — | — | — | — | — | — | — | +++ | — | — |
| Osteoporosis | — | — | — | — | — | ++ | — | +++ | ++ | + | — |
| Scoliosis | — | — | — | — | ++ | + | ++ | +++ | ++ | +++ | ++ |
| Macrocephaly | — | — | — | — | — | — | — | — | — | +++ | — |
| Microcephaly | — | — | — | — | — | + | ++ | ++ | — | — | — |
| Gingival hyperplasia | — | — | — | — | — | — | — | — | — | +++ | — |
| Corneal opacity | — | — | — | — | — | + | +++ | — | — | — | — |
| Athetoid motion | — | — | — | — | — | + | +++ | — | — | — | — |
| Alopecia | — | — | — | — | — | — | — | — | +++ | — |
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


