The hereditary disorders of cornification, or the ichthyoses, are characterized by impairment in desquamation with hyperkeratosis and/or scaling. The ichthyoses are largely distinguished by their clinical and in some cases, histologic and ultrastructural features. The discovery of the underlying molecular basis of most of the forms of ichthyosis has not only further refined classification, but has also facilitated understanding of the interactions among epidermal proteins and the role of epidermal lipids in normal epidermal function. Desquamation is the end result of proteolytic degradation of corneodesmosomes (the intercellular junctions within the stratum corneum) and is abetted by friction and cell hydration. Desquamation requires normal epidermal differentiation and depends on a gradient of pH, the presence of protease inhibitors, and the generation of hygroscopic molecules within the stratum corneum cells. Abnormalities in desquamation and differentiation usually result from abnormal corneocyte shedding (retention hyperkeratosis) or from increased epidermal cell proliferation (epidermal hyperplasia).
The epidermal barrier consists of stacked corneocytes (cells of the stratum corneum) and surrounding highly hydrophobic lipid layers (lamellae) formed by the secretion of lamellar body contents at the interface of the stratum granulosum and the stratum corneum. Most of the mutations that lead to ichthyosis affect lipid metabolism or epidermal proteins, leading to barrier dysfunction and resulting in increased transepidermal water loss and decreased water-holding capacity. Many of the features of ichthyosis are thought to be continuous compensatory attempts to restore the barrier (e.g., upregulated epidermal lipid synthesis, epidermal hyperproliferation, and inflammation). This homeostatic response likely allows survival of affected individuals.
Nonsyndromic Forms of Ichthyosis
Four major forms of nonsyndromic ichthyosis have been delineated, based largely on clinical and genetic characteristics. These are the most common forms of ichthyosis:
- 1.
Ichthyosis vulgaris, the most common ichthyosis, transmitted as an autosomal semi-dominant trait
- 2.
Recessive X-linked ichthyosis (RXLI), expressed only in males and transmitted as an X-linked recessive trait
- 3.
Keratinopathic ichthyosis, an autosomal dominant trait (most common is epidermolytic ichthyosis [EI])
- 4.
Autosomal recessive congenital ichthyosis (ARCI) (most collodion babies; the lamellar ichthyosis [LI]/congenital ichthyosiform erythroderma [CIE] spectrum).
Ichthyosis Vulgaris
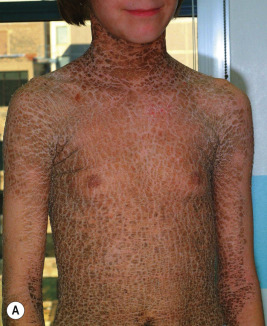
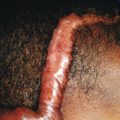
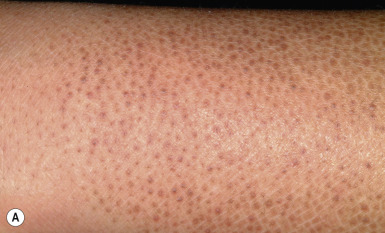
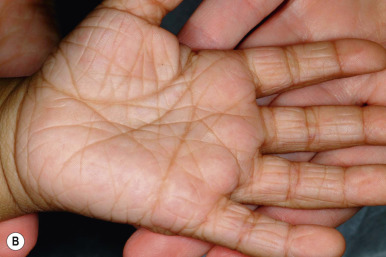
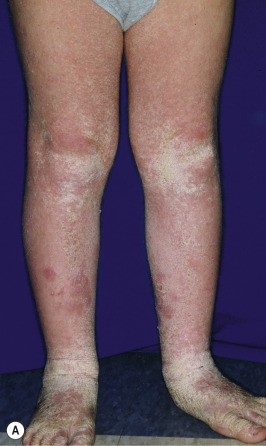
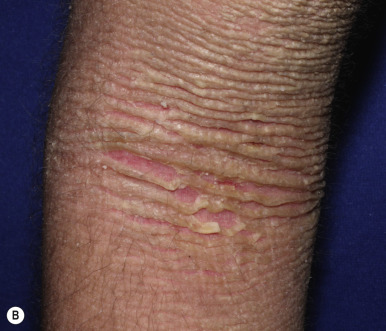
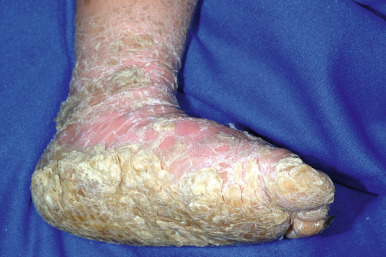
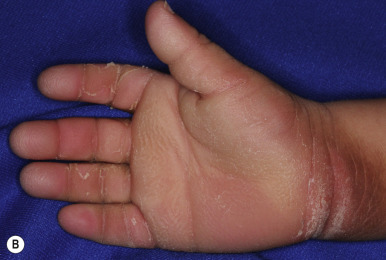
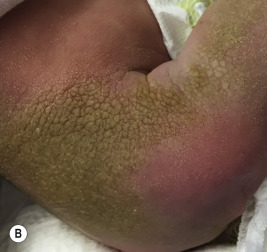
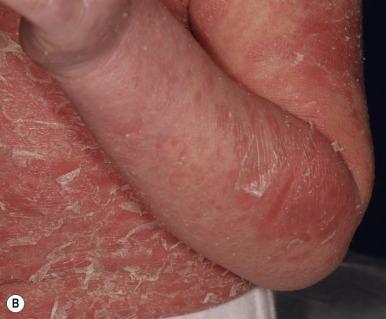
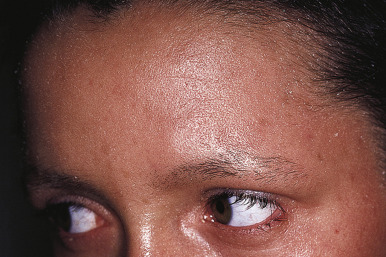
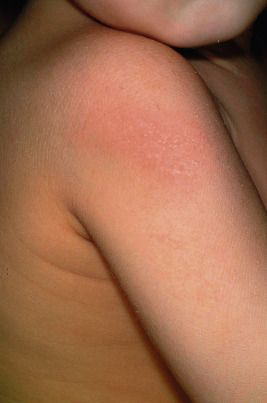
Ichthyosis vulgaris is by far the most common genetic form of ichthyosis, and the majority of individuals affected by the disorder are undiagnosed ( Table 5-1 ) either because of the minimal clinical manifestations or because the condition is dismissed as dry skin. This disorder, which is not present at birth, may be noted after the first 2 to 3 months of life and often not until later in childhood. Ichthyosis vulgaris generally improves with age, in summer, and in warm moist environments. Scales are most prominent on the extensor surfaces of the extremities and are most severe in cold and dry weather. Scales on the pretibial and lateral aspects of the lower leg are large and plate-like, resembling fish scales ( Fig. 5-1, A ); the flexural areas are characteristically spared. In other areas, small, white, bran-like scales may be seen. Scales tend to be darker on dark-skinned individuals. Scaling of the forehead and cheeks, common during childhood, generally diminishes and clears with age, but hyperlinearity and mild to moderate thickening of the palms and soles is characteristic ( Fig. 5-1, B ). Discrete hyperkeratosis may occur on the elbows, knees, and ankles.
| Ichthyosis Vulgaris | X-Linked Recessive Ichthyosis | Keratinopathic Ichthyosis, * Epidermolytic Ichthyosis | ARCI: Classic Lamellar Ichthyosis | ARCI: Classic Congenital Ichthyosiform Erythroderma | |
|---|---|---|---|---|---|
| Prevalence | 1:100 | 1:4000 males | 1:300,000 | 1:300,000 | 1:300,000 |
| Inheritance | Autosomal semi-dominant | X-linked recessive | Usually autosomal dominant | Usually autosomal recessive | Autosomal recessive |
| Onset | 2 months and beyond | 17% at birth; 83% by 1 year | Birth, with superficial blistering | Birth, as collodion baby | Birth, as collodion baby |
| Character of scales | Fine, white to larger scales, esp. on legs | Large, brown | Verrucous scale, superficial blisters | Large, plate-like scales | Fine white scaling overlying erythema |
| Localization | Can be generalized; relative sparing of flexures; hyperlinear palms | Accentuation at neck and behind ears; relative sparing of flexures | Generalized; especially at flexures and overlying joints | Generalized; ectropion, occasional alopecia, nail dystrophy | Generalized; ectropion; occasional alopecia |
| Distinct histologic features | Often shows decreased granular layer | None | Epidermolytic hyperkeratosis | Massive orthokeratosis; moderate acanthosis | More acanthosis |
| Molecular basis | Mutations in profilaggin ( FLG ); worse if both alleles | Deletions in ARSC1 (arylsulfatase C) | Mutations in KRT1 and KRT10 ; superficial form with mutations in KRT2 | Most commonly mutations in TGM1 ; rarely other types (see CIE) | Mutations in TGM1 ; ALOXE3; ALOX12B; NIPAL4; ABCA12; CYP4F22; PNPLA1; LIPN; CerS3 |
| Comments | Increased risk of atopic dermatitis and keratosis pilaris | Accumulation of cholesterol sulfate; FISH analysis to detect deletion; genital abnormalities rare; asymptomatic corneal opacities; contiguous gene deletion: with retardation, anosmia (Kallman syndrome) and/or chondrodysplasia punctata | Superficial form shows more superficial blistering and much less thickening; secondary Staphylococcus aureus infection | Transglutaminase activity can be measured in skin samples | May be associated with neurologic abnormalities |
* Previously known as epidermolytic hyperkeratosis, bullous congenital ichthyosiform erythroderma, and ichthyosis bullosa of Siemens (superficial form).
Patients with ichthyosis vulgaris often reveal an atopic background with a tendency toward atopic dermatitis, asthma, and/or allergic rhinitis. The diagnosis of ichthyosis vulgaris should be considered in patients with atopic dermatitis who show large scales, particularly on the extensor aspects of the extremities; examination of the palms and soles shows the hyperlinearity. The presence in a parent of hyperlinear palms and dry skin, especially on the lower extremities, may be helpful in confirming this diagnosis. Keratosis pilaris, which is commonly associated with ichthyosis vulgaris and atopy (see Chapter 3 , and Chapter 7 , Figs. 7-24 and 7-25 ), is most predominant on the upper arms, buttocks, and thighs.
A reduced or absent granular layer in skin sections may help to differentiate ichthyosis vulgaris from other forms of ichthyosis. Given that filaggrin is the major protein of the granular layer, it is not surprising that mutations in the gene encoding for profilaggrin, the precursor of filaggrin, are responsible. In fact, FLG mutations are observed in 7.7% of Europeans and 3.0% of Asians, although not all manifest with ichthyosis vulgaris, given the dependence on environmental factors of its phenotype. FLG mutations are uncommon in darker-skinned populations, even with ichthyosis vulgaris. Ichthyosis vulgaris is now known to be a semi-dominant condition, in contrast to the previous assumption of dominant inheritance (i.e., manifestations are seen with a mutation on one allele in 1:12 Europeans but are worse if null mutations are on both alleles (probability, 1:576). The cleaved product of profilaggrin, filaggrin, plays an important role in linking a protein (involucrin) and lipids (ceramides) in the corneocyte envelope (see Chapter 3 ). In addition, filaggrin breaks down to amino acid metabolites that increase skin hydration (natural moisturizing factor). Without filaggrin, the epidermis cannot provide normal barrier function; transepidermal water loss is increased, leading to xerosis; and the ingress of foreign substances (such as allergens and pathogens) occurs more readily, thereby increasing the risk of exposure to triggers of atopy. In fact, mutations in profilaggrin are strongly linked to the risk of atopic dermatitis and secondary asthma, regardless of the ethnicity or specific profilaggrin mutation. The risk is highest in individuals with two mutated alleles. Overall up to 30% of patients of a Northern European background with atopic dermatitis and more than 20% of Japanese with atopic dermatitis have FLG mutations. Signs of atopic dermatitis may be seen before other clinical signs of ichthyosis vulgaris, and most individuals with ichthyosis vulgaris never show evidence of atopic dermatitis. However, reduction of filaggrin expression, including in absence of FLG mutation, is a feature of autosomal dominance, because the inflammation itself and exogenous stressors suppress the expression of FLG and other important differentiation proteins, even in nonlesional skin. Emollients may improve dry skin but do not normalize the epidermal gene expression profile of filaggrin.
Individuals with FLG mutations have a generally altered risk of developing common diseases, even beyond atopic disorders. Mechanistic studies have shown increased penetration of allergens and chemicals in filaggrin-deficient skin, and epidemiological studies have found higher levels of hand eczema, irritant contact dermatitis, nickel sensitization, and serum vitamin-D levels.
Excessive exposure to factors that decrease skin barrier functions and increase the risk of atopic dermatitis should be avoided. Individuals with ichthyosis vulgaris should be protected against neonatal exposure to cats to prevent atopic dermatitis and should abstain from smoking to prevent asthma.
Although rare in children, “acquired” ichthyotic scaling has been described in patients with nutritional disorders such as hypovitaminosis or hypervitaminosis A, hypothyroidism, sarcoidosis, dermatomyositis, leprosy, tuberculosis, human immunodeficiency virus (HIV) infection, and neoplastic disorders, particularly lymphomas. It should be noted that these forms were described before the availability of testing for profilaggrin mutations; given the many undiagnosed patients, it is possible that the underlying condition served as a trigger for increased cutaneous inflammation or dryness that led to increased expressivity of the gene mutation. Pityriasis rotunda, a rare variant of acquired ichthyosis, is characterized by asymptomatic, circular or oval, brown scaly patches on the trunk or extremities. Seen primarily in individuals of Japanese, African, and West Indian origin, its occurrence in Caucasians is extremely rare. The condition may at times be associated with an underlying disorder, may follow pregnancy, or may be familial. In contrast to ichthyosis vulgaris, pityriasis rotunda is chronic, is resistant to treatment, and tends to improve only when the underlying disorder is treated.
Recessive X-Linked Ichthyosis
Recessive X-linked ichthyosis (RXLI) occurs in 1:2500 to 5000 males and results from mutation (and usually a complete deletion) in the ARSC1 gene that encodes steroid sulfatase (STS), also known as arylsulfatase C (see Table 5-1 ). The disorder has rarely been described in females who have Turner syndrome or carry the mutation on both alleles. Female carriers do not tend to show ichthyosis, because the affected gene is located at the distal tip of the X chromosome, a location that escapes X-inactivation. Thus rather than having random inactivation of one of the X chromosomes as dictated by the Lyon hypothesis, both of the alleles are expressed in every cell, providing sufficient enzyme. STS normally is concentrated in lamellar bodies and secreted into the spaces between stratum corneum cells. It degrades cholesterol sulfate, generating cholesterol for the epidermal barrier. Cholesterol sulfate itself is an epidermal protease inhibitor, so STS deficiency prevents normal degradation of the stratum corneum desmosomes and leads to corneocyte retention.
Low placental production of estrogens and elevated sulfated steroid levels have been described in the urine of mothers of boys with RXLI, associated with a difficult or prolonged labor and failure to have cervical dilation. Deletion of both ARSC1 and a contiguous gene (up to 10% of patients) results features of ichthyosis and Kallmann syndrome (associated with mental retardation, hypogonadism, and anosmia) and/or X-linked recessive chondrodysplasia punctata (bone dysplasia with stippled epiphyses).
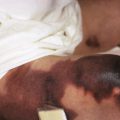
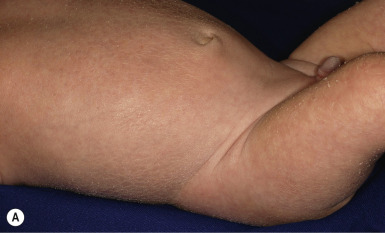
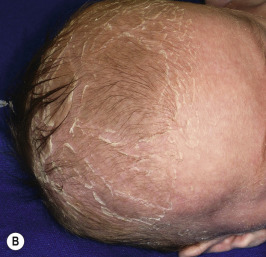
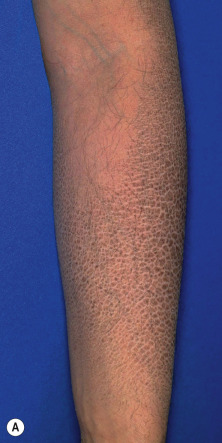
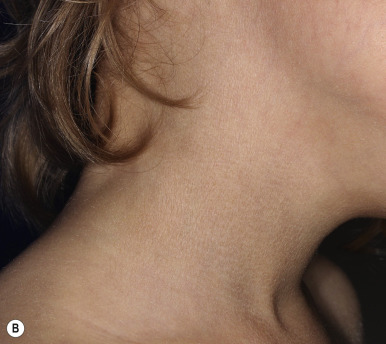
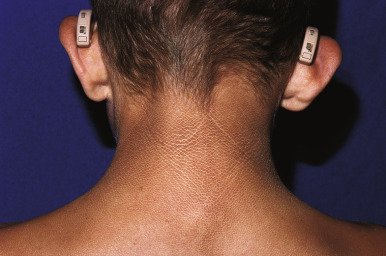
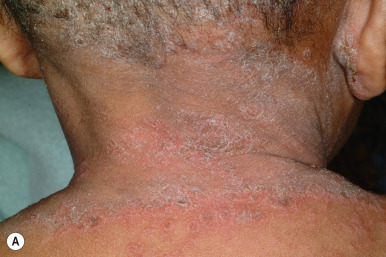
RXLI usually manifests within the first 3 months of life. Approximately 17% of affected individuals show scaling at birth, often in the form of a mild collodion-like membrane. Most develop scaling during the first 6 months of life ( Fig. 5-2 ). The severity of the scaling can range from mild to severe. This form of ichthyosis generally involves the entire body with accentuation on the scalp, neck, abdomen, back, front of the legs, and feet, but sparing the palms, soles, central face, and flexural areas ( Fig. 5-3, A ). Scales may be small to large and tend to be brown in coloration and darker in darker-skinned patients. The sides of the neck often appear dark and unwashed ( Fig. 5-3, B ). Patients may shed or molt their scales episodically, particularly in the spring and fall.
Boys with X-linked ichthyosis rarely have hypogonadism and/or cryptorchidism; testicular cancer has been described in one patient. Deep corneal opacities may be found in approximately 50% of affected adult males and less often in female carriers of this disorder. The opacities, easily detectable by slit-lamp examination, are discrete and diffusely located near the Descemet membrane or deep in the corneal stroma. Although they are a marker for the disorder in older patients, these opacities do not affect vision.
X-linked ichthyosis is often suspected prenatally, because fetal STS deficiency leads to low maternal serum and urinary estriol levels. Fluorescent in situ hybridization analysis (FISH) for the STS gene shows deletion of the gene found in 90% of patients. RXLI can also be confirmed by reduced arylsulfatase C activity in leukocytes. Elevated blood levels of cholesterol sulfate and increased mobility of β-lipoproteins have also been seen.
The features of RXLI may be seen in patients with autosomal recessive multiple sulfatase deficiency, a syndromic form of ichthyosis characterized by features of mucopolysaccharidoses, metachromatic leukodystrophy, X-linked recessive chondrodysplasia punctata, and RXLI. The disorder results from mutations in sulfatase-modifying factor 1 ( SUMF1 ), which encodes α-formylglycine generating enzyme required for posttranslation modification of sulfatases. Progressive neurologic deterioration, a feature of the metachromatic leukodystrophy, usually leads to death during infancy.
Keratinopathic Ichthyoses
The new classification of the ichthyoses has renamed the group of epidermolytic forms of ichthyosis associated with keratin gene mutations as keratinopathic. The major subgroups are epidermolytic ichthyosis (EI) (formerly called bullous CIE, Brocq type ) and superficial EI (formerly called ichthyosis bullosa of Siemen s). The designation epidermolytic hyperkeratosis is a histologic description that is not specific to this group of disorders, although it traditionally referred to EI.
Mutations are usually point mutations that lead to an abnormal but full-length keratin that incorporates into the keratin filament. The resultant keratin network functions poorly, leading to skin cell collapse and clinical blistering, especially in response to trauma. The thickening of skin is thought to be compensatory to protect against blistering but has also been linked to abnormal lamellar-body secretion. Virtually all forms are inherited in an autosomal dominant manner, although EI may rarely be autosomal recessive. EI results from mutations in KRT1 or its partner in intermediate filament formation, KRT10 ; both are expressed throughout the suprabasal layers of epidermis. Superficial EI is caused by mutations in KRT2 , which is only expressed in more superficial epidermis and also partners with keratin 10.
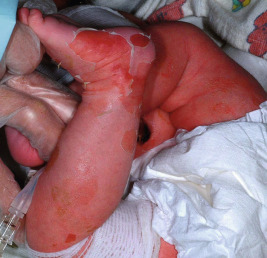
EI affects approximately 1 in 300,000 individuals, and 50% of patients have new mutations. The skin is red and may be tender at birth. Superficial bullae generally appear within the first week of life (often within a few hours after delivery; Fig. 5-4 ) and may be confused with those of epidermolysis bullosa (see Chapter 13 ) or with staphylococcal scalded skin syndrome (see Chapter 14 ). Skin thickening often appears from the third month on, but subtle thickening may be detectable during the first month of life, especially over the elbows and knees, and may be useful in suggesting the diagnosis. The blisters occur in crops and vary from 0.5 cm to several centimeters in diameter. They tend to heal quickly, consistent with their superficial location. When ruptured, they discharge clear fluid and leave raw denuded areas. Secondary bacterial infection, especially with Staphylococcus aureus , is commonly associated with this disorder.
Verruciform grayish-brown scales eventually cover most of the skin surface; the flexural creases and intertriginous areas show particularly marked involvement ( Fig. 5-5 ). Palms and soles have varying degrees of thickening and scaling ( Fig. 5-6 ), but more marked involvement often occurs in individuals with mutations in KRT1 , because keratin 9 expression in the palms and soles can compensate for abnormal keratin 10 expression, but expression of KRT1 (which is the partner for both keratins 9 and 10) remains critical. Facial involvement may occur, but ectropion does not; although scalp involvement may result in nit-like encasement of hair shafts, the hair, eyes, teeth, and nails are normal. A disagreeable body odor is commonly associated with severe forms of this disorder owing to the thick, macerated scale and overgrowth of bacteria and, less often, candida. Skin-biopsy specimens show marked hyperkeratosis with lysis of the epidermal cells above the basal cell layer (epidermolytic hyperkeratosis) leading to the bullae. Keratolytic agents are often poorly tolerated in keratinopathic forms of ichthyosis and can increase skin fragility.
Mutations in KRT1 and KRT10 can lead to other ichthyotic phenotypes as well. Mutations in either can lead to an annular variant (annular EI). Annular erythematous polycyclic scaling plaques on the trunk and extremities slowly enlarge, resolve, and later recur.
Ichthyosis Curth–Macklin (formerly called ichthyosis hystrix ) has its onset of manifestations during early childhood as progressively worsening diffuse or striate palmoplantar keratoderma (PPK) that can be associated with deep fissuring, flexural contracture, and digital constriction. Affected individuals develop characteristic “porcupine quill-like” verrucous yellow-brown scaling especially on the hands and feet and overlying the large joints. Binuclear cells and pathognomonic concentric perinuclear “shells” of aberrant keratin are characteristic ultrastructural findings. Keratin 1 mutations have been described in some but not all affected patients.
The epidermolytic hyperkeratotic form of epidermal nevus shows a histologic appearance identical to that of epidermolytic hyperkeratosis (see Chapter 9 ). This form of nevus represents a mosaic condition in which the affected skin but not the normal intervening skin carries a mutation in KRT1 or KRT10 . Individuals with more extensive forms of the epidermolytic hyperkeratotic form of epidermal nevus can have offspring with generalized epidermolytic hyperkeratosis, reflecting germline mutations. Prenatal diagnosis can be performed to in at-risk families.
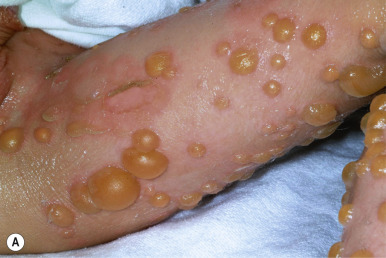
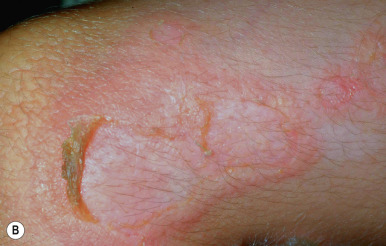
Superficial EI shows milder thickening and more superficial blistering. Palms and soles are minimally thickened if at all. Although large, tense bullae can occur intermittently ( Fig. 5-7, A ), in general the appearance of blisters has been likened to molting (Mauserung phenomenon) ( Fig. 5-7, B ) because of the superficial location of the cleavage plane. Accordingly, less hyperkeratosis is seen in sections of skin biopsies, and the lysed areas of epidermis only begin halfway up the stratum spinosum. Affected individuals may be misdiagnosed as mild EI clinically, but the limited localization of the epidermolysis histologically and the finding of mutations in KRT2 by molecular genetic testing can distinguish the disorders.
Ichthyosis en confetti , also called congenital reticular ichthyosiform erythroderma or ichthyosis variegata, is a rare autosomal recessive keratinopathy that resembles nonbullous CIE but progressively shows reversion to normal-appearing skin in a “confetti” pattern of macules. Mutations occur in the sixth or seventh exon of KRT1 or KRT10 , which seem to be predisposed to postzygotic translocations that allow protein expression of the mutated allele and correct the phenotype. The keratin is produced but stays intracellular and is not able to function normally. In some patients this reversion starts during the first years of life and is progressive, especially with keratin 1 mutations, whereas in others the onset is later during childhood or even young adulthood, especially with keratin 10 mutation. Other major features include malformation of ears, hypoplasia of mammillae, and dorsal acral hypertrichosis.
Collodion Baby
Collodion baby is a descriptive term for an infant who is born with membrane-like covering resembling collodion ( Fig. 5-8 ). The collodion baby is not a disease entity but is a phenotype common to several forms of ichthyosis. At least 65% of collodion babies have ARCI (see Autosomal Recessive Congenital Ichthyosis section), a group of genetically distinct forms of ichthyosis with overlapping clinical features ; some infants in this group (5% to 6%) shed their collodion membranes and show apparently normal skin (self-healing collodion baby [SHCB]). Less often, collodion babies shed their membranes and show features of syndromic forms of ichthyosis (Conradi–Hünermann–Happle syndrome, trichothiodystrophy [see Chapter 7 ], or RXLI). Using a new severity score for newborns with a collodion membrane, classic nonsyndromic ichthyoses were found to have higher severity scores. Recently, a collodion baby was born to a mother who received infliximab before and throughout the course of the pregnancy; although the collodion membrane shed and the skin appeared normal 1 year after birth, genotype analysis was not reported to distinguish a drug effect versus a gene mutation to cause the ichthyosis.
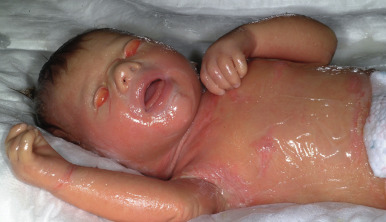
Collodion babies are often born prematurely. At birth they are completely covered by a cellophane or oiled parchment-like “membrane” that, owing to its tautness, may distort the facial features and extremities. Thus peripheral edema with digital constriction, flattened ears, and bilateral eversion of the eyelids, lips, and at times the vulva often cause affected infants to resemble one another during the first few days of life. Among the problems facing these infants are an inability to suck properly, respiratory difficulty because of restriction of chest expansion by the thick membrane, cutaneous and systemic infection, and aspiration pneumonia. Despite the thickening of the stratum corneum, this membrane is a poor barrier, leading to excessive transcutaneous fluid and electrolyte loss, hypernatremic dehydration, increased metabolic requirements, and temperature instability.
Supportive care is of primary importance in the management of collodion babies. They are best managed in a humidified incubator with special attention given to the prevention of temperature instability, sepsis, and fluid and electrolyte imbalance. Systemic antibiotic therapy should be initiated if infection is detected but not prophylactically. Desquamation is encouraged by the application of emollients rather than manual debridement; given the poor cutaneous barrier and potential toxicity, use of keratolytic agents should be avoided during the first several months of life.
Autosomal Recessive Congenital Ichthyosis
Autosomal recessive congenital ichthyosis (ARCI) encompasses a wide range of clinical phenotypes that range from classic harlequin ichthyosis (HI) and nonbullous CIE to classic LI. The same individual may show a range of overlapping clinical features; for example, retinoid treatment may decrease the lamellar scaling but increase erythroderma. Failure to thrive is a feature of ARCI, especially if severe, and short stature may ensue. Overall, ARCI occurs in approximately 1 in 100,000 to 300,000 live births. The diagnosis is based on clinical findings; biopsy is not helpful unless needed to exclude alternative diagnoses. The features of the ARCI group of disorders usually persist throughout the affected individual’s lifetime. Many affected individuals complain of associated pruritus. Owing to the obstruction of eccrine glands by the overlying hyperkeratosis, severely affected patients tend to experience hyperpyrexia, heat intolerance, difficulty with perspiration, and heat exhaustion during periods of warm or hot, humid weather and vigorous physical exercise. Squamous cell carcinomas have been reported at a younger age.
Mutations in nine known genes have been shown to result in the ARCI phenotype, and most of these mutated genes have been related to both LI and CIE phenotypes (see Table 5-1 ). A few cases of autosomal dominant LI have been described. The discovery of the underlying genetic basis in families with ARCI has facilitated the prenatal diagnosis based on genotyping rather than the riskier diagnosis by fetal skin biopsy. Up to 55% of individuals with ARCI have mutations in the gene encoding transglutaminase 1 ( TGM1 ), particularly patients with the LI phenotype. TGM1 crosslinks several proteins to form the cornified envelope surrounding corneocytes. In patients missing transglutaminase, transglutaminase activity is undetectable in frozen skin specimens. HI has been shown to result from nonsense mutations in the gene encoding the ABCA12 transporter. Deficiency of ABCA12 leads to perturbation of lipid transport, which then leads to a paucity of lamellar bodies, the upper epidermal lamellar structures that provide intracellular lipids to the stratum corneum, and to premature terminal differentiation of keratinocytes. ABCA12 has also been shown to be important for protease function. Mutations in ABCA12 that lead to the LI or more commonly CIE phenotype tend to be missense mutations, so that some gene product is present for function, leading to the milder phenotype. Other genes found to be mutated in ARCI encoding include lipoxygenase-3 ( ALOXE3 ), 12(R)-lipoxygenase ( ALOX12B ), ichthyin ( NIPAL4 ) CYP4F22, lipase N ( LIPN ), PNPLA1, and ceramide synthase 3 ( CERS3 ). Some of these encode proteins of the hepoxilin pathway ( ALOX12B, ALOXE3, and NIPAL4 ). In addition to their disruption of stratum corneum lipid synthesis, these enzymes (or the ichthyin receptor) within the lipoxygenase pathway may also disrupt the processing of profilaggrin to filaggrin.
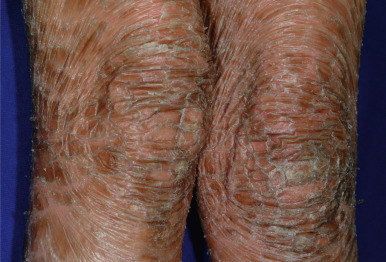
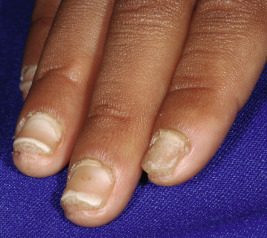
The clinical features of the classic LI phenotype may range from very mild to severe. Individuals who show the greatest severity of LI have large lamellar plate-like scales with relatively mild underlying erythroderma, ectropion (eversion of an edge or margin of the eyelid resulting in exposure of the palpebral conjunctiva) ( Fig. 5-9 ), and mild eclabium (eversion of the lips) (see Table 5-1 ). Lamellar scales are large, quadrangular, yellow to brown-black, often thick, and centrally adherent with raised edges resembling armor plates (hence the term LI ) ( Fig. 5-10 ). Scales are most prominent over the face, trunk, and extremities, with a predilection for the flexor areas. Cheeks are often red, taut, and shiny; more scales appear on the forehead than on the lower portion of the face. The palms and soles are almost always affected in LI; severity varies from increased palmar markings to a thick keratoderma with fissuring. The scalp is often scaly with scarring partial hair loss (especially with TGM1 mutations). Involvement of the nails is variable. They may be stippled, pitted, ridged, or thickened, often with marked subungual hyperkeratosis.
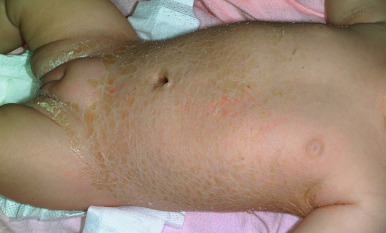
Variant forms of LI from mutations in TGM1 manifest in a more limited distribution of lesional skin. Patients with bathing suit ichthyosis (BSI) are born with a full collodion membrane and transition to LI, but within the first months of life the scaling on the extremities clears ( Fig. 5-11 ). The residual LI on warmer skin areas (axillae, trunk, scalp, neck) has been linked to temperature-sensitive mutations in TGM1 . Mutations in TGM1 that encode proteins sensitive to hydrostatic pressure can result in the SHCB. Affected neonates show either a generalized or acral collodion membrane at birth that clears entirely as the baby transitions to the dry environment postnatally. The TGM1 mutations in both BSI and SHCB phenotypes are missense mutations that are predominantly in the catalytic core domains. ALOX12B and ALOX3B mutations have also been described with SHCB.
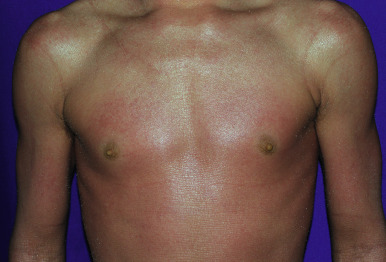
Classic nonbullous CIE is characterized by a much more prominent erythrodermic component that may first become apparent as the collodion membrane is shed; some patients show CIE at birth without a classic collodion membrane. Affected individuals show fine white scales on the face, scalp, and trunk, although scaling may be more plate-like scales on the extensor surfaces of the legs ( Fig. 5-12 ). The degree of ectropion is variable but often milder than with LI, and there is less PPK. Cicatricial alopecia is possible, and nails may show thickening and ridging. Some patients with CIE show intrauterine growth retardation and/or failure to thrive, although nutritional deficiency and gastrointestinal abnormalities are uncommon. Patients with CIE may have associated neurologic abnormalities.
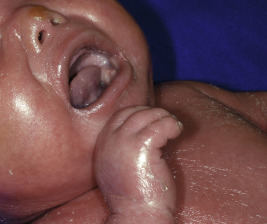
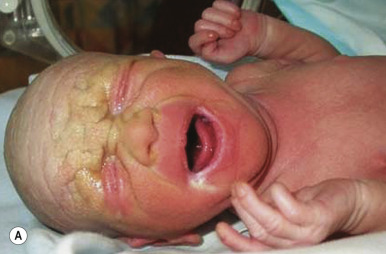
HI is the most severe form of ARCI. At birth, the disorder manifests as profoundly thickened, armor-like skin that is fissured into polygonal, triangular, or diamond-shaped plaques that simulate the traditional costume of a harlequin ( Fig. 5-13 ). Rigidity of the skin results in marked ectropion; everted O -shaped lips with a gaping fishmouth deformity; and a distorted, flattened, and undeveloped appearance to the nose and ears. The skin rigidity can restrict respiratory movements, sucking, and swallowing. The hands and feet are ischemic, hard, and waxy, often with poorly developed digits and an associated rigid and claw-like appearance. Flexion deformity of the limb joints is common, and the nails may be hypoplastic or absent. Restrictive dermopathy (see Chapter 6 ) shows congenital contractures, tight skin, ectropion, and intrauterine growth retardation and can thus sometimes be confused with HI, but it shows no hyperkeratosis or scaling.
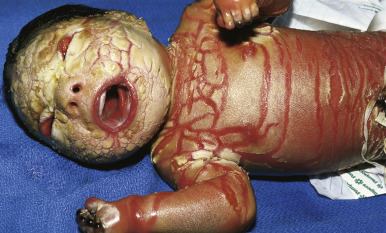
Death is usually associated with prematurity, pulmonary infection (in part associated with hypoventilation caused by thoracic rigidity), poor feeding, excessive fluid loss, poor temperature regulation, or sepsis as a result of cutaneous infection. The severity may be variable, however, and prolonged survival has been achieved by intensive supportive measures, emollients and, in some cases, oral administration of systemic retinoids. Initiation of aggressive intervention for babies with HI is controversial. Even with administration of systemic retinoids or spontaneous clearance of the armor-like scaling, the optimal outcome resembles that of severe CIE ( Fig. 5-14 ). Physicians often reserve initiation for babies who survive the first few weeks, because most infants are stillborn or die during the neonatal period (usually during the first few hours or days of life); however, it is unclear whether use of systemic retinoids leads to a better outcome than excellent supportive care, especially during the neonatal period therapy. Pulmonary issues can be ongoing in infants and young children. Prenatal diagnosis of HI has been suspected based on ultrasound-based discovery of distal arthrogryposis and can be performed definitively by molecular analysis.
Other Forms of Nonsyndromic Ichthyosis
Loricrin Keratoderma
Loricrin keratoderma (also called Camisa disease, a variant of Vohwinkel keratoderma) is an autosomal dominant disorder with ichthyosis and PPK. Mutations occur in the gene encoding loricrin, a protein that is linked by transglutaminase-1 to involucrin and other proteins of the corneocyte envelope, thereby participating in barrier function and normal epidermal maturation. Affected individuals may be born with a collodion membrane and later show a mild, nonerythrodermic generalized ichthyosis with flexural accentuation. The PPK is initially noted during the first weeks of age, is often transgrediens, and may show a honeycomb pattern that resembles that of Vohwinkel syndrome (connexin defect). Pseudoainhum (constricting bands of the digits) may occur but usually not until adolescence or even adulthood; the starfish-shaped keratoses of Vohwinkel syndrome are not seen with loricrin keratoderma. Alopecia is occasionally seen but not other ectodermal abnormalities or hearing impairment. Parakeratosis on routine skin biopsy is a characteristic histologic feature, but termination mutations in the C-terminus have been linked to milder PPK without pseudoainhum and without parakeratosis on affected skin biopsy.
Erythrokeratodermia Variabilis and Progressive Symmetric Erythrokeratodermia
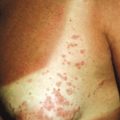
Erythrokeratodermia variabilis (EKV) is a dominantly inherited ichthyosis characterized by two distinct types of lesions: (1) sharply marginated, pruritic, or burning areas of erythema with finer scaling that are often figurate in configuration and undergo changes in size, shape, and distribution during a period of days to weeks; and (2) hyperkeratotic plaques with thick, yellow-brown scales that usually overlie erythema ( Fig. 5-15 ). Lesions are most often symmetrically distributed on the limbs, trunk, and buttock with relative sparing of the face, scalp, and flexures. In contrast to the chronic but remitting appearance of these plaques and figurate lesions, plaques on the knees, elbows, Achilles tendons, and soles of the feet are often persistent. PPK has been described in 50% of affected families.
Although lesions are usually noted at birth or shortly thereafter during the first year of life, in a few individuals the onset has been noted during late childhood or early adulthood. The disorder may partially regress at puberty and tends to improve in summer. Patients usually tend to respond well to systemically administered retinoids.
EKV has been known to result from mutations in genes that both map to chromosome 1p35.1 and encode interacting connexins: GJB3 encoding connexin 31 and GJB4 gene encoding connexin 30.3. Most recently, EKV with hyperpigmented, well-delineated keratotic plaques was shown to result from mutations in GJA1 encoding connexin 43 ( Fig. 5-16 ). Individuals with symmetrical EKV lesions during early infancy that clear by 25 years of age may develop progressive spinocerebellar ataxia in adulthood in association with heterozygous mutations in ELOVL4. ELOVL4 encodes an elongase needed for synthesis of very long-chain fatty acids in skin, eyes, and the neurologic system. When ELOVL4 mutations affect both alleles, ichthyosis can occur in association with spastic diplegia and severe neurodevelopmental delay, Homozygous ELOVL4 mutations must be distinguished from Sjögren–Larsson syndrome (see Sjögren–Larsson Syndrome section).
Progressive symmetric erythrokeratodermia (PSEK; Darier–Gottron syndrome) is a dominant disorder that tends to have its onset during infancy, tends to stabilize after 1 or 2 years, and may partially regress at puberty. PSEK has been distinguished from EKV by the absence of the migrating red patches, the typical sparing of the chest and abdomen with scaling plaques limited to the extremities, buttocks, and face, and a higher incidence of PPK (≈50% of cases); however, the conditions overlap phenotypically, and patients have been described with features of both, suggesting that PSEK and EKV are a single disorder with variable manifestations. In fact, a G12D missense mutation in connexin 30.3 has been shown to cause both PSEK and EKV.
Peeling Skin Syndrome
Peeling skin syndrome (PSS), or keratolysis exfoliativa congenita, is an unusual autosomal recessive disease characterized by life-long, spontaneous superficial peeling of the skin that may be persistent or periodic. The Nikolsky sign tends to be positive. The desquamation has been associated with increased stratum corneum and serum kallikrein levels. Two generalized types have been described. Desquamation is generalized, other than the palms and soles, which may be mildly thickened. Seasonal variation with worsening during summer months has been described.
In type A, the onset may be at birth but commonly begins in early childhood (by 6 years of age). These patients are asymptomatic. Skin biopsy shows a thickened stratum corneum with an intracorneal or subcorneal separation. Type A is caused by mutations in CHST8, which encodes a Golgi transmembrane N-acetylgalactosamine-4-O-sulfotransferase (GalNAc4-ST1). In type B, the condition always begins at birth. Patients show erythematous migratory peeling patches and complain of associated pruritus or burning. Patients with the type B form may show short stature and easily removable anagen hairs. Biopsy specimens reveal psoriasiform thickening of the epidermis and a subcorneal or intracorneal split. Type B results from loss of corneodesmin, found in the stratum corneum and hair follicle.
An acral form of hereditary PSS is characterized by life-long painless peeling of predominantly the dorsal aspect of the hands and feet in superficial sheets and exacerbation during summer months with increased sweating. A facial variant has been described. This acral form is caused by mutations in TGM5 encoding transglutaminase-5, which crosslinks epidermal proteins. Acral PSS in association with plantar keratoderma and peeling can be associated with mutations in encoding cystatin A ( CSTA ), a protease of the cornified cell envelope. Cystatin A mutations can also be associated with generalized mild scaling and exfoliation. Acral PSS may be misdiagnosed as localized epidermolysis bullosa simplex.
Keratosis Linearis-Ichthyosis Congenital-Keratoderma
Keratosis linearis-ichthyosis congenital-keratoderma (KLICK) is a rare, autosomal recessive disorder characterized by distinctive striate hyperkeratosis in the flexures (perpendicular to the fold) and PPK. It results from a single-nucleotide deletion in the 5′ untranslated regions (UTRs) of the proteasome maturation protein ( POMP ) gene, which reduces levels of POMP and leads to proteasome insufficiency, increased endoplasmic reticulum (ER) stress, and perturbed processing of profilaggrin in differentiating keratinocytes.
Syndromic Forms of Ichthyosis
Syndromic forms of ichthyosis may be associated with a variety of extracutaneous abnormalities, most commonly involving the hair (e.g., Netherton; ichthyosis follicularis, alopecia, and photophobia [IFAP] and ichthyosis, hypotrichosis, and sclerosing cholangitis [IHSC] syndromes and ichthyosis with hypotrichosis; trichothiodystrophy) and neurologic system (e.g., Sjögren–Larsson; Refsum; dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma [CEDNIK]; and mental retardation, enteropathy, deafness, neuropathy, ichthyosis, and keratoderma [MEDNIK] syndromes). Some syndromic disorders are lethal in the neonate (e.g., Neu–Laxova syndrome), infant (e.g., Gaucher disease type 2; arthrogryposis, renal dysfunction, and cholestasis [ARC] syndrome; multiple sulfatase deficiency) or child (e.g., CEDNIK syndrome).
Neutral Lipid Storage Disease with Ichthyosis
Neutral lipid storage disease with ichthyosis, or Chanarin–Dorfman syndrome, is a rare autosomal recessive disorder seen primarily in individuals of Middle Eastern or Mediterranean descent. The clinical phenotype can variably include liver steatosis with hepatomegaly, muscle weakness/myopathy, ataxia, neurosensory hearing loss, subcapsular cataracts, nystagmus, strabismus, and mental retardation, but ichthyosis of the CIE phenotype is an almost constant finding. Patients are often born as collodion babies, occasionally with ectropion and eclabium. Skin biopsies show skin thickening with foamy keratinocyte cytoplasm owing to prominent neutral lipid droplets in the basal cells and eccrine glands seen best on oil-red O-stained frozen sections. Serum lipids are normal, although the triglyceride content of lymphocytes, macrophages, and fibroblasts in culture is two to 20 times that of normal cells. Muscle and liver enzymes may be elevated twofold to threefold. The diagnosis is confirmed by a peripheral blood smear, which shows lipid droplets in granulocytes (Jordan anomaly) that are also seen in the leukocytes of heterozygous carriers of Chanarin–Dorfman syndrome. Mutations have been identified in ABHD5 or CGI-58 , a gene that encodes an enzyme expressed during differentiation in lipid transporting lamellar granules of epidermis that is required for triglyceride degradation and normal barrier function. A diet of moderate amounts of carbohydrates and medium-chain triglycerides, together with oral acitretin 0.5 mg/kg per day, has been shown to improve both the CIE and liver function tests.
CHIME Syndrome
Individuals with CHIME syndrome (also called Zunich neuroectodermal syndrome ) show a combination of coloboma, heart defects, ichthyosiform dermatosis, mental retardation, and ear anomalies, including conductive hearing loss. The disorder is caused by mutations in PIGL, a de-N-acetylase required to make glycosylphosphatidylinositol (GPI) anchors, which are membrane glycolipids that anchor the C-terminus of proteins during posttranslational modification. The skin is notably thickened and dry at birth, with pruritus often developing during the first months of life ( Fig. 5-17 ). The colobomas are usually retinal, although choroidal colobomas have been described. Several heart defects, including pulmonic stenosis, ventricular septal defect, transposition of the great vessels, and tetralogy of Fallot, have been associated. Patients show a typical facies with hypertelorism; a broad, flat nasal root; upslanting palpebral fissures; epicanthic folds; a long columella but short philtrum; macrostomia; full lips; and cupped ears with rolled helices. All patients show brachydactyly. The hair may be fine and sparse, and trichorrhexis nodosa has occasionally been described. Some patients have renal or urologic anomalies, and cleft palate has been described in association.
KID Syndrome
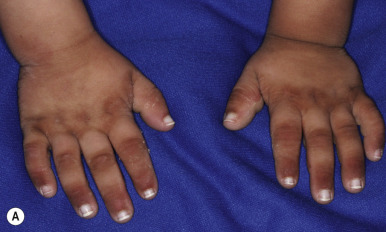
KID syndrome is a rare autosomal dominant disorder characterized by keratitis, congenital ichthyosis, and neurosensory deafness. Patients are usually born with erythematous skin that progressively becomes more thickened and leathery during the first months of life. Generalized tiny stippled papules are characteristic ( Fig. 5-18 ), and 90% of patients develop well-defined verrucous plaques, especially on the face and limbs. Alopecia may be congenital (25%) and ranges from sparse hair to total alopecia ( Fig. 5-19 ); a thick yellow scale may cover the scalp at birth. Most patients show PPK with a stippled or leathery pattern. Nails tend to be dystrophic, and sweating may be diminished.
The hearing loss is congenital, neurosensory, and nonprogressive; it can be detected by brainstem auditory-evoked potential testing. In contrast, ocular features are rarely seen at birth but progress and become evident by childhood or early adolescence. Photophobia may be the earliest sign, and the characteristic corneal vascularization and keratoconjunctivitis sicca lead to pannus formation and marked reduction in visual acuity. KID syndrome must be distinguished from IFAP syndrome (see Chapter 7 ), an ichthyotic condition in which patients have total alopecia, thickened skin with spiny projections, PPK, and photophobia with decreased visual acuity.
Almost half of patients with KID syndrome have recurrent bacterial and candidal infections of the skin, eyes, and ear canals. Some patients have demonstrated abnormal chemotaxis and impaired lymphocyte proliferative responses to Candida albicans . The follicular occlusion syndrome (including hidradenitis suppurativa) has been described in some patients, may lead to scarring alopecia, and may require surgical intervention. More than 10% of patients develop squamous cell carcinoma of the skin, especially acral, or tongue occasionally during childhood, and trichilemmal tumors have also been described. Dandy–Walker malformation has been described in several affected individuals.
The disorder results from mutations in one of GJB2 or GJB6, encoding connexins (proteins that create channels critical in intercellular communication) 26 and 30, respectively. The latter connexin is also mutated in patients with Clouston syndrome, which shares the alopecia, nail dystrophy, PPK, and sometimes photophobia of KID syndrome (see Chapter 7 ). A baby with KID syndrome from a connexin 26 mutation has recently been described whose unaffected mother had the same point mutation but had an additional, more distal null mutation in the same allele that protected her from having phenotypic manifestations; the second mutation was lost in the baby, presumably by prezygotic reversion during meiosis of the maternal gamete.
Therapy of KID syndrome is largely supportive. Chronic administration of fluconazole has improved the verrucous plaques of cutaneous candidiasis. Cochlear implants and corneal transplants have been used to correct the sensorineural hearing loss and corneal vascularization, respectively. Recent research has suggested that mefloquine, a US Food and Drug Administration (FDA)-approved antimalarial drug, inhibits the aberrant connexin 26 hemichannels in keratinocytes from a mouse model of KID syndrome and may be a future intervention.
Netherton Syndrome
Netherton syndrome (NS) is an autosomal recessive condition that combines ichthyosis, atopy, and hair shaft deformities. NS presents during the neonatal or early infantile period with generalized scaling erythroderma but not a true collodion-baby phenotype. Neonates with NS are usually born prematurely and develop the eruption in utero or during the first weeks of life. Failure to thrive is often profound, requiring hospitalization for nutritional support and correction of the hypernatremic dehydration that may be associated.
Patients may have diarrhea associated with intestinal villus atrophy, and the majority experience sepsis, upper and lower respiratory infections, and cutaneous S. aureus infection. Adults with NS are also at risk for extensive papillomavirus infection, usually involving the genital region, and may develop squamous cell carcinoma. A variety of immunologic abnormalities have been described, suggesting that NS should be considered a primary immunodeficiency disorder. These include reduced memory B cells, defective response to vaccination, impaired antibody amplification and class switching, decreased natural killer (NK)-cell cytotoxicity, a skewed T-helper (Th) 1 phenotype, and increased proinflammatory cytokine levels. Treatment with intravenous immunoglobulin may cause an increase in NK cell cytotoxicity and clinical improvement.
Ichthyosis linearis circumflexa, the characteristic skin change associated with NS, is characterized by migratory, polycyclic scaly lesions with a peripheral double-edged scale ( Fig. 5-20 ). Although most commonly seen in association with NS, patients may show only the ichthyosis linearis circumflexa without the hair shaft abnormalities or other features of NS. Ichthyosis linearis circumflexa is not generally seen before 2 years of age and occurs eventually in only 70% of patients. The ichthyosiform erythroderma that is the typical manifestation in the neonatal and infantile periods tends to improve with increasing age. Partial remissions have been noted and spontaneous fluctuation is common, but there is little tendency to spontaneous resolution. Routine histologic examination of skin-biopsy sections is not helpful, but immunohistochemical studies show absent or reduced expression of LEKTI, and electron microscopic studies have revealed features that are specific to NS.
The classic hair shaft abnormality, trichorrhexis invaginata (“bamboo hairs,” “ball-and-socket deformity”), is thought to result from a defect in keratinization of the internal root sheath. The hair defect results in easy hair breakage and hair that is poorly manageable, dry, and lusterless ( Fig. 5-21 ) (see Chapter 7 , Fig. 7-5 ). Multiple hairs from different areas should be examined, because only 20% to 50% of hairs may be affected. Examination of hairs from the eyebrow region often is most fruitful, and dermoscopy facilitates visualization of the “matchstick” hair defect. Finding the hair shaft disorder is particularly difficult in the affected neonate.
NS results from loss-of-function mutations in SPINK5 , which encodes lymphoepithelial Kazal-type-related inhibitor ( LETKI ), a serine protease inhibitor. The increase in serine protease activity (kallikrein 5) leads to decreases in desmosomal proteins (desmoglein 1 and desmocollin 1) with premature degradation of corneocyte desmosomes and excessive desquamation. Approximately two-thirds of patients show pruritic atopic-like dermatitis, food allergies, urticaria, angioedema, asthma, and/or anaphylaxis. In addition, most patients have increased levels of circulating eosinophils and immunoglobulin (Ig) E. These atopic manifestations likely result from the unregulated kallikrein 5 activity, which activates PAR2 and TSLP (as in atopic dermatitis, see Chapter 3 ) as well as other cytokines and contributes to both the very poor skin barrier and the cutaneous inflammation. The marked impairment in barrier function can lead to significant absorption of topically applied medication, necessitating careful monitoring of serum levels or adrenal suppression. Application of hydrocortisone 1% ointment caused Cushing syndrome in an 11-year-old boy, and immunosuppressive serum levels may be detectable after application of tacrolimus ointment. Nevertheless, several patients have responded well to application of topical calcineurin inhibitors without detectable absorption.
NS should be distinguished from other forms of ichthyosis, especially with abnormal hair, barrier defects, and early failure to thrive ( Table 5-2 ). These include severe dermatitis, multiple allergies, and metabolic wasting (SAM) syndrome ; epidermal growth factor receptor (EGFR) deficiency ; and ADAM17 deficiency. Alopecia with ichthyosis is also a feature of trichothiodystrophy (see Chapter 7 ), ichthyosis hypotrichosis syndrome, and IHSC syndrome (see Table 5-2 ).
| Disorder | Inheritance | Gene | Features Resembling Netherton Syndrome | Differentiation from Netherton Syndrome |
|---|---|---|---|---|
| SAM syndrome | AR Loss-of-function mutations | DSG1 | CIE with PPK, no collodion membrane; failure to thrive; hypernatremia; barrier defect; dermatitis; high IgE; malabsorption; eosinophilic esophagitis; multiple food allergies; recurrent infections; hypotrichosis; hypoalbuminemia | May have microcephaly, growth hormone deficiency, developmental delay, cardiac defects; psoriasiform dermatitis with acantholysis in skin sections; absence of desmoglein |
| ADAM17 deficiency | AR Loss-of-function mutations | ADAM17 | Psoriasiform erythroderma/widespread pustules; failure to thrive; malabsorption; short, broken hair; recurrent infections | Bloody diarrhea; cardiomyopathy/cardiomyositis |
| EGFR deficiency | AR Loss-of-function mutations | EGFR | Erythema, scaling, and widespread pustules; alopecia; failure to thrive, watery diarrhea, high IgE and eosinophils, hypernatremia, hypoalbuminemia; recurrent bronchiolitis | Cardiovascular issues |
| Trichothiodystrophy | AR Loss-of-function mutations | ERCC2, ERCC3, GTF2H5, C7Orf11 (see Chapter 7 ) | CIE-like ichthyosis; short, brittle hair; “tiger tail” hair shaft defect under polarized microscopy | May have impaired intelligence, decreased fertility, short stature, and photosensitivity |
| IHS (also called autosomal recessive ichthyosis with hypotrichosis [ARIH] syndrome) | AR Loss-of-function mutations | ST14 (encodes matriptase); abnormal filaggrin processing | Generalized, congenital ichthyosis with sparing of face, palms, and soles; diffuse nonscarring alopecia of scalp, eyelashes, and eyebrows from birth, but improves to sparse, unruly hair during adolescence and merely recession of the frontal hair line by adulthood | May have patchy follicular atrophoderma and hypohidrosis; photophobia from corneal abnormalities; blepharitis; dental abnormalities; hair microscopy may show pili torti or pili bifurcati |
| IHSC (or NISCH) syndrome | AR Loss-of-function mutations | CLDN1 (encodes claudin 1, structural protein of tight junctions) | Congenital generalized scaling, predominantly on the limbs and abdomen and sparing skinfolds, palms and soles; coarse, curly hair with frontotemporal cicatricial alopecia | Congenital paucity of bile ducts or sclerosing cholangitis leads to neonatal jaundice with hepatomegaly; oligodontia, and enamel dysplasia; blood smears show small eosinophils and keratinocyte vacuoles without lipid contents |
* Netherton syndrome must also be distinguished from severe atopic dermatitis and immunodeficiency disorders.
Treatment of NS is largely supportive, although administration of infliximab led to sustained improvement and decreased thymic stromal lymphopoietin levels, and narrow-band ultraviolet B (UVB) light has led to marked cutaneous improvement within a few months. A Phase 1 ex vivo gene therapy trial is underway in the United Kingdome, with the normal SPINK5 gene introduced by lentivirus into the grafted patient keratinocytes.
Refsum Disease
Refsum disease is an autosomal recessive neurocutaneous disorder caused by deficiency in oxidation of phytanic acid, a branched, long-chain fatty acid derived from dietary chlorophyll. The clinical features usually develop in late childhood or early adult life and progress slowly during months to several years. Neurologic manifestations are most prominent and include sensorineural deafness, anosmia, failing vision, night blindness owing to retinitis pigmentosa and a progressive weakness, foot drop, and loss of balance caused by a mixed sensorimotor neuropathy and cerebellar involvement. Delayed diagnosis may result in severe neurologic impairment, wasting, and depression. The associated ichthyosis, which can either coincide with or postdate the neurologic features, resembles ichthyosis vulgaris or in severe untreated cases, LI. Accentuated palmoplantar markings are associated.
Refsum disease results from mutations in one of two genes, either PHYH (also named PAHX ), which encodes the peroxisomal enzyme phytanoyl-CoA hydroxylase, or PEX7, which encodes the PTS2 (peroxisomal targeting signal 2) receptor. Phytanic acid cannot be synthesized by humans and is mainly derived from plant chlorophyll. Normally serum levels are undetectably low but in Refsum disease may account for up to 30% of serum lipids. The accumulation of phytanic acid disturbs the cholesterol balance and may alter lipid degradation. Histology shows variably sized vacuoles in the epidermal basal and suprabasal cells, corresponding to the lipid accumulation seen with lipid stains of frozen sections. The diagnosis is based on the demonstration of increased levels of phytanic acid in the patient’s serum, tissue, or urine. Therapy consists of a chlorophyll-free diet and avoidance of phytanic acid-containing foods.
Sjögren–Larsson Syndrome
The ichthyosis of the autosomal recessive disorder, Sjögren–Larsson syndrome (SLS), usually manifests in the neonatal period as fine, white scaling, accentuated in flexural areas. Erythema is occasionally present at birth but clears within months. Presentation as a collodion baby is rare. By 1 year of age, the ichthyosis of SLS is not erythrodermic but shows generalized velvety lamellar thickening (often with a yellowish hue) particularly on the trunk and neck with minimal desquamation ( Fig. 5-22 ), PPK, and relative sparing of the face. The skin is characteristically pruritic, and most patients are hypohidrotic. The degree of scaling varies from mild to severe ( Fig. 5-23 ) and does not change with increasing age. Hair and nails are normal. The neurologic disease usually presents within the first year with failure to reach normal developmental milestones and the onset of spasticity. Phenotypic variability is seen, and some patients have been described with mild neurologic features of SLS without associated skin disease. Spasticity and muscle paresis is most pronounced in the legs, and most affected persons become dependent on a wheelchair for mobility. Most patients have a learning disability to a variable degree and a speech disorder, and some show seizures, short stature, kyphosis, and enamel hypoplasia. The pathognomonic retinal “glistening dots” are not present in all patients, but photophobia is common.

SLS results from mutations in the fatty aldehyde dehydrogenase gene ( FALDH or ALDH3A2 ), a component of fatty alcohol, fatty alcohol oxidoreductase (FAO), which converts fatty alcohol to fatty acid. Epidermal cells in affected individuals show abnormal lamellar bodies and lipid droplets, consistent with defective lipid metabolism. Prenatal diagnosis of SLS is possible by measurement of FAO activity in cultured amniocytes or chorionic cells, histologic analysis, and/or analysis of fetal deoxyribonucleic acid (DNA) if the gene defect is known. The ichthyosis is treated with topical keratolytic agents and retinoids ; dietary supplementation with medium-chain fatty acids is generally not helpful. The leukotriene inhibitor zileuton may decrease the associated pruritus, but no therapy to date has been found to slow the progressive neurologic deterioration. Ichthyosis with more severe neurologic changes than SLS (spastic diplegia, seizures, and intellectual disability) can also result from biallelic mutations in ELOVL4, a fatty acid elongase that catalyzes the first and rate-limiting step in very long chain fatty acid synthesis (see Erythrokeratodermia Variabilis and Progressive Symmetric Erythrokeratodermia section).
Ichthyosis Prematurity Syndrome
Ichthyosis prematurity syndrome (IPS) is an autosomal recessive disorder in which affected babies are born more than 6 weeks prematurely in association with polyhydramnios and opaque amniotic fluid because of the extensive shedding of epidermal cells. Neonates often have perinatal respiratory distress and sometimes life-threatening neonatal asphyxia of amniotic keratin debris. Typically babies are born with generalized thick spongy desquamating skin that resembles vernix caseosa, accentuated on the scalp and eyebrows. During the neonatal period the scaling may resemble cobblestones overlying moderate erythroderma. Although the marked thickening clears in survivors, xerosis with follicular keratosis and pruritus persists, and patients often show atopic dermatitis, dermographism, asthma, and eosinophilia. More than 70% develop respiratory and/or food allergy. Mutations have been identified in SLC27A4, the fatty-acid transport protein 4 ( FATP4 ) gene, which encodes a fatty-acid transporter and leads to defective stratum corneum lipid homeostasis. FATP4 may associate with ichthyin and transglutaminase 1 in lipid processing essential for maintaining the epidermal barrier function.
Neu–Laxova Syndrome
Another autosomal recessive form of congenital ichthyosis associated with a high risk of early death is Neu–Laxova syndrome, characterized by severe intrauterine growth retardation, an edematous appearance, lung hypoplasia, microcephaly, and abnormal brain development with lissencephaly and agenesis of the corpus callosum. The ichthyosis tends to be present at birth but ranges in severity from mild ichthyosis to an HI appearance. Patients tend to show typical facies, including protuberant eyes with a flattened nose, slanted forehead, micrognathia, deformed ears, and a short neck. Some affected neonates show microphthalmia or cleft palate. Syndactyly, limb or digital hypoplasia, and limb contractures are common, and X-rays often show poor bone mineralization. Mutations occur in one of three genes of the L-serine biosynthesis pathway, phosphoglycerate dehydrogenase ( PHGDH ), phosphoserine aminotransferase ( PSAT1 ) and phosphoserine phosphatase ( PSPH ). Neu–Laxova represents the severe end of the spectrum of disorders caused by mutations in these genes (others have variable neurologic manifestations). Craniofacial and limb defects have been seen in a variety of syndromes with reduced intrauterine movement (fetal akinesia/hypokinesis sequence).
Gaucher Syndrome Type 2
Gaucher syndrome type 2 results from an absence of lysosomal β-glucocerebrosidase, which hydrolyzes glucosylceramide to ceramide. The neonate with Gaucher syndrome type 2 (acute infantile cerebral form) may be a collodion baby with the onset during infancy of neurologic signs and hepatosplenomegaly. Glucosylceramide and ceramide are critical components of the intercellular bilayers of the stratum corneum and play a role in epidermal barrier function. The absence of glucocerebrosidase leads to abnormal skin thickening and increased transepidermal water loss. Even when the skin appears normal clinically in affected patients, ultrastructural abnormalities in lamellar membranes may be seen. Despite enzyme replacement therapy, nonneurologic manifestations of Gaucher disease may progress.
CEDNIK Syndrome
CEDNIK syndrome first manifests between 5 and 11 months of age with progressive neurologic deterioration. Affected infants show a generalized mild LI phenotype with sparing of skin folds but palmoplantar thickening. The hair tends to be fine and sparse. Microcephaly, neuropathy, cerebral dysgenesis, sensorineural deafness, optic nerve atrophy, neurogenic muscle atrophy, and cachexia are associated, and affected individuals usually die within the first decade of life. Patients with CEDNIK syndrome have mutations in SNAP29 , a component of the secretory (SNARE [soluble NSF attachment protein receptor]) pathway that is important for vesicle fusion and lamellar granule maturation and secretion. In CEDNIK syndrome, glucosylceramide and kallikrein-containing granules are abnormally retained in the stratum corneum, leading to retention hyperkeratosis and an abnormal epidermal barrier.
MEDNIK Syndrome
MEDNIK syndrome is an autosomal recessive disorder that shows manifestations at birth or within the first weeks of life. Among the ichthyoses, it most closely resembles EKV and in fact has also been called EKV3. Nail thickening and mucosal involvement may be associated. Patients show congenital sensorineural deafness, psychomotor and growth retardation, mental retardation, and peripheral neuropathy. The severe, congenital, chronic diarrhea is life-threatening. MEDNIK syndrome results from mutations in AP1S1 , encoding a subunit (1A) of an adapter protein complex (AP-1) that is involved in the organization and transport of proteins during skin and spinal-cord development.
ARC Syndrome
ARC syndrome presents with generalized desquamative lamellar scaling within the first days to weeks of life but not at birth. Ectropion and mild scarring alopecia may be present. The distinguishing features are the associated arthrogryposis (contractures of the limbs, particularly of the knee, hip and wrist; rocker bottom feet; talipes equinovarus), renal tubular degeneration with metabolic acidosis, and intrahepatic bile-duct hypoplasia with cholestasis. Patients may also show cerebral malformations, hypothyroidism, deafness, dysmorphic features, and large, dysfunctional platelets. Death ensues during the first year of life. ARC syndrome results from mutations in VPS33B, which regulates SNARE protein-mediated fusion of membrane vesicles required for lamellar body secretion. Incomplete ARC syndrome has been described without the arthrogryposis.
Conradi–Hünermann–Happle Syndrome
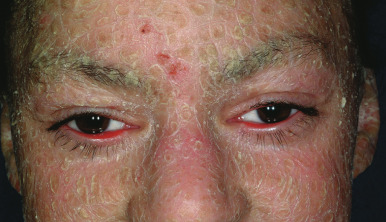
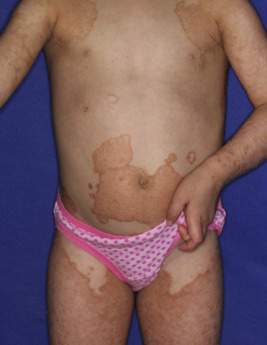
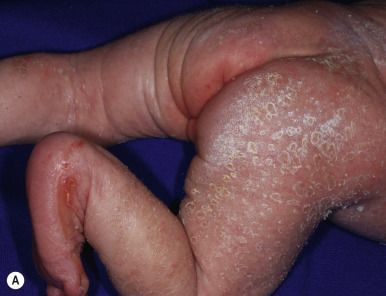
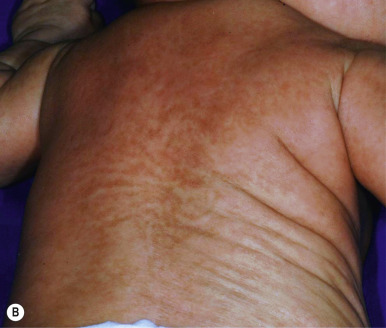
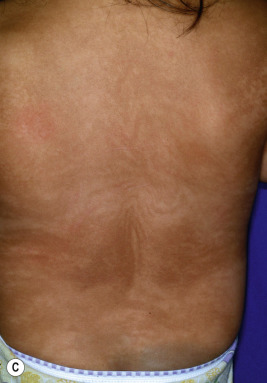
The key clinical features of Conradi–Hünermann–Happle (CHH) syndrome (also called X-linked dominant chondrodysplasia punctata type II ) are linear ichthyosis, chondrodysplasia punctata, cataracts, and short stature. Neonates tend to show severe ichthyosiform erythroderma with patterned yellowish markedly hyperkeratotic plaques ( Fig. 5-24, A ). After the first 3 to 6 months of life the erythroderma and scaling resolve, leaving erythema and later follicular atrophoderma in a distribution that follows Blaschko lines, hypopigmentation and hyperpigmented streaks particularly on the trunk ( Fig. 5-24, B and C ), and circumscribed cicatricial alopecia of the scalp and eyebrows ( Fig. 5-25 ). Patients may show persistent psoriasiform lesions in intertriginous areas (ptychotropism) ( Fig. 5-26 ). In addition to the cutaneous findings, patients with CHH show asymmetric skeletal involvement with punctate calcification of the epiphyseal regions that usually results in an asymmetric shortening of the long bones (especially the humeri and femora) and sometimes in severe kyphoscoliosis, facial dysplasia, and hip dislocation. Unilateral or bilateral sectorial cataracts; patchy, coarse, lusterless hair; nasal bone dysplasia with saddle-nose deformity (see Fig. 5-25 ), and a high-arched palate further characterize the disease. The bony abnormalities but not the skin lesions of CHH syndrome have been described in association with teratogenic exposures to medications and infections and maternal autoimmune diseases.
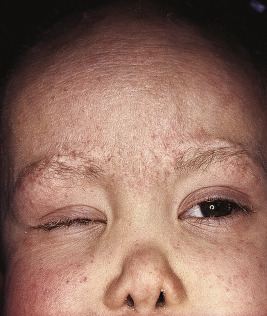
X-chromosomal inactivation explains the distribution of skin lesions along Blaschko lines, the sectorial cataracts, and the asymmetric skeletal abnormalities. Abnormal cholesterol synthesis/metabolism has been detected in patients with CHH, and mutations have been identified in emopamil binding protein (EBP), which encodes 3β-hydroxysteroid- Δ8, Δ7-isomerase, a key component in cholesterol biosynthesis. Abnormal lamellar granules and malformed intercellular lipid layers have been detected ultrastructurally, and plasma sterol analysis shows markedly elevated levels of 8(9)-cholesterol and 8-dehydrocholesterol.
CHILD Syndrome
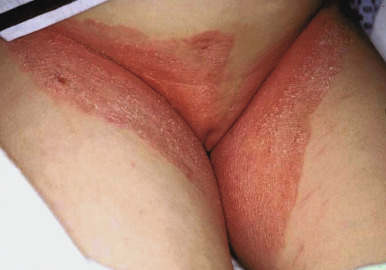
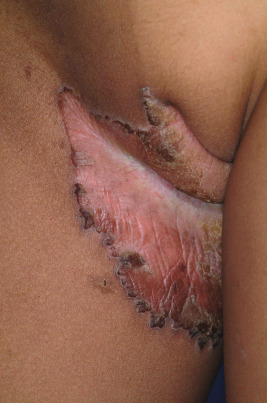
The CHILD syndrome is a congenital disorder characterized by congenital hemidysplasia, ichthyosiform erythroderma, and limb defects. Also known as unilateral CIE, the hallmark of the disorder is the sharp midline demarcation and its largely unilateral cutaneous and skeletal features ( Fig. 5-27 ). This X-linked dominant condition occurs almost exclusively in girls and is presumed to be lethal in affected males. The only case in a boy is thought to represent early postzygotic mosaicism. The inflammatory ichthyosiform skin lesions of CHILD syndrome may be present at birth or develop during the first few months of life. They are characterized by yellow and waxy scaling and/or streaks of inflammation and scaling that is often patchy but may follow Blaschko lines. Similarly, streaks of normal skin may be interspersed within the area of the CHILD nevus. Unilateral alopecia and severe nail dystrophy with claw-like nails have been described. The face is typically spared. With increasing age, lesions may improve or even clear spontaneously, but lesions in intertriginous areas (ptychotropism) tend to persist and be the most severely affected sites. Rarely a localized hyperkeratotic plaque with a sharp demarcation at the midline is the sole manifestation of the CHILD syndrome (CHILD nevus) ( Fig. 5-28 ). A mild form of CHILD syndrome has been described in three generations, suggesting genetic control of the skewing of X inactivation.
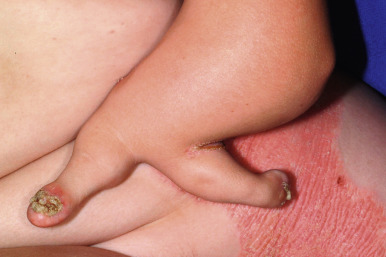
Ipsilateral skeletal hypoplasia, ranging in severity from hypoplasia of the fingers to complete agenesis of an extremity, is an important feature of CHILD syndrome. As with the skin changes, unilaterality is not absolute, and slight changes may be present on the contralateral side. The molecular basis for the unilaterality is not understood, but the epidermal changes correlate with mutant gene expression in the epidermis; leukocytes show skewing of X-chromosome inactivation, and virtually all circulating cells express the normal gene. Punctate epiphyseal calcifications may be demonstrable by radiography but tend to disappear after the first few years of life. Cardiovascular and renal abnormalities are the major visceral manifestations of CHILD syndrome, although anomalies of other viscera have been described. Biopsy of skin lesions shows epidermal thickening with characteristic infiltration of the papillary dermis of histiocytes showing foamy cytoplasm (verruciform xanthoma). Inactivating mutations have been identified in the NSDHL gene encoding a 3β-hydroxysteroid dehydrogenase, which functions upstream of EBP in the cholesterol synthesis pathway. Treatment with keratolytic agents and retinoids is poorly tolerated, but topical application of compounded 2% lovastatin/2% cholesterol leads to dramatic improvement.
Treatment of Lesional Skin of Nonsyndromic and Syndromic Ichthyoses
Treatment of patients with most forms of ichthyosis involves topical application of keratolytic agents and topical or systemic administration of retinoids. The Foundation for Ichthyosis and Related Skin Types (FIRST; www.firstskinfoundation.org ) is a support group for patients and families with disorders of cornification. In additional to educational materials, FIRST provides information about commercially available treatment options. Several other foundations worldwide support families with ichthyosis and have educational websites as well (e.g., www.ichthyosis.org.uk and www.ictiosis.org ).
The management of all types of ichthyosis consists of retardation of water loss, rehydration and softening of the stratum corneum, and alleviation of scaliness and associated pruritus. Daily to twice-daily baths using a superfatted soap or a soapless cleanser followed immediately by application of the emollient to moist skin can be helpful for all forms. Shorter baths are preferred for patients with ichthyosis vulgaris, especially with associated atopic dermatitis. However, many patients with LI or EI have found long baths to be particularly helpful. Ichthyosis vulgaris and RXLI can be managed quite well by topical application of emollients and the use of keratolytic agents to facilitate removal of scales from the skin surface. α-Hydroxy acid preparations such as lactic and glycolic acids are the most commonly used as agents to desquamate excessive scale and increase hydration. Urea in concentrations of 10% to 20% has a softening and moisturizing effect on the stratum corneum and is helpful in the control of dry skin and pruritus. Salicylic acid is another effective keratolytic agent and can be compounded into petrolatum at concentrations between 3% and 6% to promote shedding of scales and softening of the stratum corneum. When it is used to cover large surface areas for prolonged periods, however, patients should be monitored for salicylate toxicity, most commonly complaints of tinnitus. The combination of 6% salicylic acid in propylene glycol may be particularly helpful for keratoderma of the palms and soles, especially when used under occlusive wraps. The LI phenotype of ARCI generally requires more potent keratolytic agents; individuals with milder forms of LI often respond well to the topical application of tazarotene ( Fig. 5-29 ). As the skin normalizes, the risk of irritation from tazarotene increases, sometimes leading to dermatitis and then requiring therapy to be intermittent. Topical N-acetylcysteine with or without urea (e.g., 10% N-acetylcysteine in 5% urea) has also been used for LI. Recombinant transglutaminase-1 enzyme encapsulated in liposomes reversed the ichthyosis phenotype and barrier function in a skin-humanized mouse model of LI.