The genetic basis for common diseases such as atopic dermatitis and psoriasis, in addition to rare diseases, has been partially elucidated. Some genetic disorders are explained by mutations in a specific gene or genes that lead to a specific clinical phenotype that can be recognized based in part on the skin manifestations. These are termed genodermatoses, and recognizing the skin features can lead to earlier diagnosis and management, potentially preventing disease. Genetic disorders are often grouped into three categories: chromosomal, single gene, and polygenic. The advancement of genetic testing has allowed the discovery of postzygotic mutations that only affect a small area of the body. Many segmental patterns of congenital lesions are due to postzygotic mutations. These postzygotic mutations leading to congenital lesions of the vascular system and epidermal nevi are discussed in their respective chapters. Chromosomal disorders can be numeric, such as trisomy and monosomy, or structural, resulting from translocations or deletions. Most genodermatoses show single-gene or mendelian inheritance (autosomal dominant, autosomal recessive, or X-linked recessive genes). Polygenetic syndromes often involve complex interactions of genes. Occasionally a second genetic hit is required to manifest skin findings.
Autosomal dominant conditions require only a single gene to produce a given phenotype. Usually the patient has one affected parent or is affected by a new mutation. The disease is transmitted from generation to generation. Autosomal recessive traits require a homozygous state to produce the abnormality. The pedigree may reveal parental consanguinity. Parents will be clinically unaffected or have mild manifestations but often have affected relatives. X-linked conditions occur when the mutated gene is carried on the X chromosome. If a disease is X-linked recessive, the loss is evident in males (XY), who do not have a second X chromosome to express the normal allele. Therefore X-linked recessive traits occur almost exclusively in males. They cannot transmit the disease to sons (who inherit their Y chromosome), but all their daughters will be carriers. Carrier females who are heterozygous (having one normal and one abnormal X chromosome) occasionally show some evidence of the disease. This occurs as a result of lyonization, the physiologic inactivation of one of the X chromosomes. X-linked dominant disease states are usually lethal in males. Survival is possible in females who retain a normal allele. Because the mutation is lethal in many affected cell lines, females typically demonstrate loss of normal tissue in the affected segments (narrow Blaschko segments, loss of digits, microphthalmia, loss of teeth). X-linked dominant traits result in pedigrees in which more than one female is affected but no males express the disease. Rarely, males may survive, especially if they have Klinefelter syndrome (XXY) or a postzygotic mutation leading to mosaicism such that only some cells are affected.
Mosaicism is the presence of two or more genetically distinct cell lines in a single individual. It may occur as a result of physiologic inactivation of one X chromosome (lyonization) or as the result of a postzygotic somatic mutation. Mosaicism often presents in a linear and whorled pattern along the lines of Blaschko. In mosaic states, genes that are detrimental to a cell population during fetal development (e.g., incontinentia pigmenti) typically result in thin segments because they are overgrown by the adjacent normal tissue. Conversely, genes that confer a growth advantage during fetal development (e.g., mutated tumor suppressor gene in segmental neurofibromatosis) may result in broad, plaque-type lesions that have grown beyond the boundaries of a typical Blaschko segment.
In autosomal dominant conditions, a normal allele remains but is not enough to prevent disease. Loss of heterozygosity (LOH) is a second genetic mutation leading to a defect in the remaining normal allele that explains why a lesion is only in a limited part of the skin. LOH may give rise to segments of the body with an exaggerated presentation of the syndrome. The affected area corresponds to a Blaschko segment or plaque. The forehead plaque of tuberous sclerosis is related to a mutation in a tumor suppressor gene. The loss of the tumor suppressor gene imparts a growth advantage, and LOH leaves no suppressor gene product in the segment. As a result, the affected segment grows beyond its Blaschko boundaries, forming a broad plaque.
When a patient presents with segmental distribution of a disorder, it is critical to determine whether the disorder is a result of mosaicism or LOH. In LOH, the abnormal allele is present throughout the body, including gonadal tissue. In a patient who presents with segmental neurofibromatosis but has Lisch nodules or axillary freckling, LOH rather than mosaicism is likely to account for the segmental presentation. The risk of passing the gene to a child is about 50/50. A genetic counselor should be involved to discuss risk of transmission to offspring because the mechanisms may be complex. Patients with mosaicism based on postzygotic somatic gene mutation may have gonadal mosaicism and may be capable of passing on the gene. Gonadal mosaicism is more likely when more than one segment is present on different regions of the body. Before gastrulation, when a cavity forms in the embryo, every cell is pluripotent and can give rise to an entire organism, or it can contribute to multiple sites of the body. At gastrulation, cells become dedicated to produce specific segments of the body. Blaschko segments in different regions suggest a mutation that occurred before gastrulation, when the involved cell lines could contribute to different parts of the body, including the gonads. Polygenetic disorders, such as psoriasis, may also present with limited and linear forms that may relate to segmental LOH or postzygotic mutation.
Online Mendelian Inheritance in Man ( OMIM.org ) contains a comprehensive database of known genetic disorders and has a search function that allows the clinician to match clinical manifestations with possible diagnoses. When a patient has more than one very unusual finding OMIM can help determine whether the patient may have a genetic disorder linking the findings. PubMed’s clinical query function can also be used to match manifestations with syndromes, and Genetest.org lists sources for genetic testing.
Happle R: Superimposed segmental manifestation of polygenic skin disorders. J Am Acad Dermatol 2007; 57: 690.
X-linked, mosaic and related disroders with abnormal pigment
Incontinentia Pigmenti
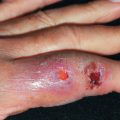
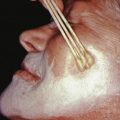
Also known as Bloch-Sulzberger disease, incontinentia pigmenti is an X-linked dominant condition characterized by whorled pigmentation on the trunk, preceded by vesicular and verrucous changes. It appears in girls during the first weeks after birth ( Fig. 27.1 ). Most lesions are evident by the time the infant is 4–6 weeks old. A vesicular phase is present in nearly all cases. This first stage begins in most individuals before 6 weeks of age and is replaced by verrucous lesions after several weeks to months in two thirds of patients. Although these usually resolve by 1 year of age, lesions may persist or recurr for many years. In the third, or pigmentary, phase, pigmented macules in streaks, sprays, splatters, and whorls follow the lines of Blaschko. The pigmentary stage may last for many years and then fade away, leaving no sequelae. The stages are not mutually exclusive. Therefore it is not uncommon for a young child to have some areas still in a verrucous stage while others are hyperpigmented. A fourth stage may be seen in some adult women, manifesting subtle, faint, hypochromic or atrophic linear lesions, most often on the extremities.
Histologically, the vesicular stage is characterized by spongiosis with eosinophils. As the lesions mature, clusters of dyskeratotic cells appear within the epidermis. Dyskeratotic cells predominate in the verrucous stage, and pigment incontinence (dermal melanophages) predominates in hyperpigmented lesions.
Other cutaneous changes include patchy alopecia at the vertex of the scalp, atrophic changes simulating acrodermatitis chronica atrophicans on the hands, onychodystrophy, late subungual tumors that resemble subungual keratoacanthoma and may have underlying lytic bone lesions, and palmoplantar hyperhidrosis. Extracutaneous manifestations occur in 70%–90% of patients. Most frequently involved are the teeth (up to 90%), bones (40%), central nervous system (CNS; 33%), and eyes (35%). Immune dysfunction with defective neutrophil chemotaxis and elevated immunoglobulin E (IgE) has been reported. Eosinophilia is common. Incontinentia pigmenti is an important cause of neonatal seizures and encephalopathy.
Dental abnormalities usually manifest by the time the individual is 2 years old. Dental defects may be the only stigmata still present in the mother and thus can be helpful in the diagnosis of an affected child. Dental defects include delayed eruption, partial anodontia (43%), microdontia, and cone- or peg-shaped teeth (30%). The most common CNS findings are seizures in approximately 10% followed by mental retardation, spastic paralysis, microcephaly, destructive encephalopathy, and motor impairment. The eye changes include strabismus, cataracts, retinal detachments, optic atrophy, vitreous hemorrhage, blue sclerae, and exudative chorioretinitis. Skeletal abnormalities include syndactyly, skull deformities, dwarfism, spina bifida, clubfoot (talipes), supernumerary ribs, hemiatrophy, and shortening of the legs and arms. Rarely, tricuspid insufficiency, right ventricular hypertrophy, and pulmonary hypertension have been described.
Incontinentia pigmenti is caused by a mutation in the nuclear factor-κB ( NEMO) gene on the X chromosome, localized to Xq28. Therefore it is typically passed from mother to daughter although there are reports of germline mosaic males giving birth to affected daughters. The gene is generally lethal in male fetuses, although males with Klinefelter syndrome (47,XXY) may survive. Mosaicism may also account for some cases in males. NEMO mutations also cause X-linked ectodermal dysplasia with immunodeficiency, characterized by alopecia, hypohidrosis, dental anomalies, and defects in humoral immunity. Osteopetrosis and lymphedema may be present.
Incontinentia pigmenti achromians is a term that has been replaced with the term pigmentary mosaicism (see section on pigmentary mosaicism ). Mendelian susceptibility to mycobacterial disease is a rare syndrome predisposing to infection with weakly virulent mycobacteria, such as Mycobacterium avium complex, Mycobacterium bovis, bacille Calmette-Guérin (BCG), and environmental nontuberculous mycobacteria. The causative mutations in NEMO selectively affect the CD40-dependent induction of interleukin-12 (IL-12) in mononuclear cells. These patients typically clinically manifest with hypohidrotic ectodermal dysplasia or rarely incontinentia pigmenti.
Use of ruby lasers to treat pigmented lesions in infants and young children may worsen the condition. Usually, the end stage of streaks of incontinentia pigmenti start to fade at age 2 years, and by adulthood, there may be minimal residual pigmentation.
Naegeli-Franceschetti-Jadassohn Syndrome
Naegeli-Franceschetti-Jadassohn syndrome differs from incontinentia pigmenti in that the pigmentation is reticular, with no preceding inflammatory changes, vesiculation, or verrucous lesions. Vasomotor changes and hypohidrosis are present. There is reticulate pigmentation involving the neck, flexural skin, and perioral and periorbital areas. Diffuse keratoderma and punctiform accentuation of the palms and soles may occur. Dermatoglyphics are abnormal, producing atrophic or absent ridges on fingerprints. Congenital malalignment of the great toenails may be found. Dental abnormalities are common, and many patients are edentulous. Both genders are equally affected, and the syndrome appears to be transmitted as an autosomal dominant trait related to mutations in keratin 14, causing increased susceptibility to tumor necrosis factor (TNF)-α–induced apoptosis. The syndrome is allelic to dermatopathia pigmentosa reticularis (DPR), which manifests with similar cutaneous findings but DPR patients have absent dermatoglyphs.
Bustamante J, et al: Genetic lessons learned from X-linked mendelian susceptibility to mycobacterial diseases. Ann NY Acad Sci 2011; 1246: 92.
Fusco F, et al: Incontinentia pigmenti. Orphanet J Rare Dis 2014; 9: 93.
Fusco F, et al: Unusual father-to-daughter transmission of incontinentia pigmenti due to mosaicism in IP males. Pediatrics 2017; 140: e20162950.
Itin PH, et al: Spontaneous fading of reticular pigmentation in Naegeli-Franceschetti-Jadassohn syndrome. Dermatology 2010; 221: 135.
Mahmoud BH, et al: Controversies over subungual tumors in incontinentia pigmenti. Dermatol Surg 2014; 40: 1157.
Marques GF, et al: Incontinentia pigmenti or Bloch-Sulzberger syndrome. An Bras Dermatol 2014; 89: 486.
Minić S, et al: Systematic review of central nervous system anomalies in incontinentia pigmenti. Orphanet J Rare Dis 2013; 8: 25.
Okita M, et al: NEMO gene rearrangement (exon 4-10 deletion) and genotype-phenotype relationship in Japanese patients with incontinentia pigmenti and review of published work in Japanese patients. J Dermatol 2013; 40: 272.
Poziomczyk CS, et al: Incontinentia pigmenti. An Bras Dermatol 2014; 89: 26.
Zhang Y, et al: Incontinentia pigmenti (Bloch-Siemens syndrome). Eur J Pediatr 2013; 172: 1137.
Pigmentary Mosaicism
Pigmentary mosaicism is a term that encompasses congenital hypopigmentation and hyperpigmentation in multiple patterns that is due to alterations in genetic pathways responsible for pigmentation. Pigmentation changes can be either hypopigmentation or hyperpigmentation and are often manifested along lines of Blaschko. Some of the affected genes are also important in other tissues and thus markers of systemic disease. For example, if there are defects in neural crest cells that are programmed to form both the skin and the CNS or eye there can be manifestations in all of the tissues. Genetic mutations that occur in earlier in development lead to a more widespread pattern of pigmentation changes and likely a greater chance that some of those cells with mutations were the progenitors for other tissues, thus increasing the risk for systemic disease associations. Many patients with localized pigmentary mosaicism especially when unilateral and blocklike have no associated systemic findings (segmental pigmentary disorder). There are many historical descriptive terms for different types of pigment but because the pathophysiology is the same, the term pigmentary mosaicism is more correct. Pigmentary mosaicism can result from chromosomal abnormalities, with most demonstrating mosaicism for aneuploidy or unbalanced translocations. There is no treatment for pigmentary mosaicism although the pigment may normalize over time in some.
Pigmentary Mosaicism With Hypopigmentation
Incontinentia pigmenti achromians (IPA) and hypomelanosis of Ito (HI) are terms that have historically been used, but because most people with HI have been described as having seizures and/or developmental delay whereas many people with blaschkoid hypopigmentation do not have these manifestations, it is favored to use the term pigmentary mosaicism to avoid the assumption that children with hypopigmentation will have poor prognosis. The hypopigmentation typically follows the lines of Blaschko ( Fig. 27.2 ). If there are system findings, which may be more likely in children with more diffuse cutaneous involvement, affected individuals can have associated anomalies of the CNS, eyes, hair, teeth, skin, nails, musculoskeletal system, or internal organs, including polycystic kidney disease. Patients may manifest psychomotor or mental impairment, autism, microcephaly, coarse facies, and dysmorphic ears.
Because there are many different types of mutations that can lead to hypopigmentation, these patients should not be considered to have one syndrome but instead multiple rare syndromes many of which have not been named. Some patients have had associated Sturge-Weber syndrome–like leptomeningeal angiomatosis. A recent group of patients was described mammalian target of rapamycin (mTOR) with pigmentary mosaicism of the hypopigmented type with megalencephaly and focal cortical dysplasia due to mosaic mTOR mutations. Several patients have demonstrated trisomy 13 with hypopigmented mosaicism.
Baxter LL, et al: The etiology and molecular genetics of human pigmentation disorders. Wiley Interdiscip Rev Dev Biol 2013; 2: 379.
Hogeling MI, Frieden IJ: Segmental pigmentation disorder. Br J Dermatol 2010; 162: 1337.
Pigmentary Mosaicism With Hyperpigmentation
Linear and whorled nevoid hypermelanosis is the term historically used to describe blaschkoid distribution of hyperpigmented streaks without preceding bullae or verrucous lesions. Sparing of mucous membranes, eyes, palms, and soles is noted. Congenital anomalies, such as mental retardation, cerebral palsy, atrial septal defects, dextrocardia, auricular atresia, hemiatrophy, and patent ductus arteriosus, may be present but seem to be less prevalent with hyperpigmentation than with hypopigmented mosaicism. Bilateral giant cerebral aneurysms have been reported. Biopsy of pigmented areas demonstrates increased pigmentation of the basal layer and prominence of melanocytes without incontinence of pigment. Again, patients who have localized pigmentation especially in blocklike unilateral patterns seem to be less likely to have systemic manifestations. Most cases appear to be sporadic, although familial cases have been reported. The differential diagnosis includes other causes of blaschkoid hyperpigmentation such as incontinentia pigmenti, verrucous epidermal nevi that have not proliferated yet, and McCune-Albright syndrome.
Cohen J 3rd, et al: Analysis of 36 cases of Blaschkoid dyspigmentation. Pediatr Dermatol 2014; 31: 471.
Mehta V, et al: Linear and whorled nevoid hypermelanosis. Int J Dermatol 2011; 50: 491.
Metta AK, et al: Linear and whorled nevoid hypermelanosis in three successive generations. Indian J Dermatol Venereol Leprol 2011; 77: 403.
Disorders Due to Sex Chromosomes
X-Linked Reticulate Pigmentation Disorder
X-linked reticulate pigmentation disorder with systemic manifestations that presents with reticulate hyperpigmentation due a mutation that leads to amyloid buildup. In males, cutaneous involvement is characterized by reticulate hyperpigmentation of the skin, characteristic facies, and severe systemic involvement. In the carrier females, manifestations are limited to the skin.
Anderson RC, et al: X-linked reticulate pigmentary disorder with systemic manifestations. Pediatr Dermatol 2005; 22: 122.
Klinefelter Syndrome
Klinefelter syndrome, the most common sex chromosome disorder, consists of hypogonadism, gynecomastia, eunuchoidism, small or absent testicles, and elevated gonadotropins. The patient may have a low frontal hairline, sparse body hair with only a few hairs in the axillary and pubic areas, scanty or absent facial hair in men, and shortening of the fifth digit of both hands.
Thrombophlebitis and recurrent or chronic leg ulcerations may be a presenting manifestation; these may be more common than previously reported. The cause of the hypercoagulable state is believed to be an increase in plasminogen activator inhibitor 1 levels. Patients are at an increased risk of lupus erythematosus and a variety of cancers, especially male breast cancer, hematologic malignancies, and sarcomas (retinoblastoma and rhabdomyosarcoma).
Many of these patients are tall; some are obese. Psychiatric disorders occur in about one third of patients and patients can have decreased mental capacity. Klinefelter syndrome is most frequently associated with an XXY sex chromosome pattern, although other variations occur as the number of X chromosomes increases. Androgen therapy may result in improvements in appearance and function.
XXYY Genotype
The XXYY genotype is considered to be a variant of Klinefelter syndrome. In addition to the changes seen in Klinefelter, vascular changes occur in XXYY patients, such as cutaneous angiomas, acrocyanosis, and peripheral vascular disease leading to stasis dermatitis. Hypertelorism, clinodactyly, pes planus, and dental abnormalities are common. Systemic manifestations include asthma, cardiac defects, radioulnar synostosis, inguinal hernia, cryptorchidism, CNS defects, attention-deficit disorder, autism, and seizures.
XYY Genotype
Patients with an XYY karyotype can have very severe acne with onset in childhood and be more aggressive and have cognitive delays, although these studies were performed on prisoners so there may be a selection bias.
Turner Syndrome
Turner syndrome, also known as gonadal dysgenesis, is characterized by a webbed neck, low posterior hairline margin, increased carrying angle at the elbow (cubitus valgus), congenital lymphedema, and a triangular mouth. Patients may demonstrate alopecia of the frontal area on the scalp, koilonychia, cutis laxa, cutis hyperelastica, mental retardation, short stature, infantilism, impaired sexual development, primary amenorrhea, numerous melanocytic nevi, angiokeratomas, and an increased risk of melanoma, pilomatricoma, and thyroid disease. Coarctation of the aorta is frequently found. There may be an increased incidence of alopecia areata and halo nevi in these patients.
Patients with Turner syndrome are females who have only one X chromosome. Mosaic Turner syndrome exists in which only some cells are missing the other X. Loss of long-arm material (Xq) can result in short stature and ovarian failure, but deletions distal to Xq21 do not appear to affect stature. Loss of the short arm (Xp) produces the full phenotype. Patients with very distal Xp deletions usually have normal ovarian function.
No specific treatment is available for Turner syndrome. Human growth hormone (hGH) has been used to treat the short stature. A review of the Cochrane Central Register of Controlled Trials determined that hGH increases short-term growth, but few data exist regarding its effects on final height.
Multiple pterygium syndrome (Escobar syndrome) is a rare autosomal recessive disorder characterized by multiple congenital joint contractures and multiple skin webs that may mimic Turner syndrome.
Balsera AM, et al: Distinct mechanism of formation of the 48, XXYY karyotype. Mol Cytogenet 2013; 6: 25.
Castelo-Branco C: Management of Turner syndrome in adult life and beyond. Maturitas 2014; 79: 471.
Dillon S, et al: Klinefelter’s syndrome (47,XXY) among men with systemic lupus erythematosus. Acta Paediatr 2011; 100: 819.
Güven A, et al: Multiple pterygium syndrome. J Pediatr Endocrinol Metab 2011; 24: 1089.
Kasparis C et al: Childhood acne in a boy with XYY syndrome. BMJ Case Rep 2014 Jan 6; 2014.
Rogol AD, et al: Considerations for androgen therapy in children and adolescents with Klinefelter syndrome (47, XXY). Pediatr Endocrinol Rev 2010; 8: 145.
Rasopathies
The Ras-Map kinase pathway (including Raf, ERK, and MEK) is critical for cellular growth. Many different genetic disorders involving mutations in the genes controlling various parts of this pathway present with similar features. Historically as these diseases were described they had significant clinical overlap and this is due to the defects involving the same pathway. These disorders are now grouped under the category of RASopathies.
Neurofibromatosis Type 1
The four types of neurofibromatosis are neurocutaneous syndromes that are manifested by developmental changes in the nervous system, bones, and skin.
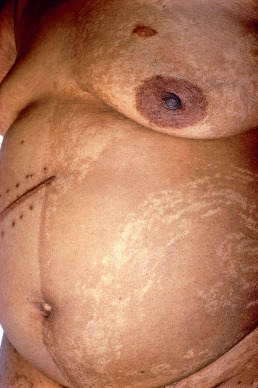
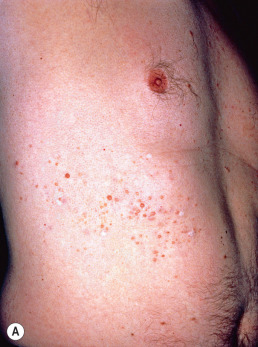
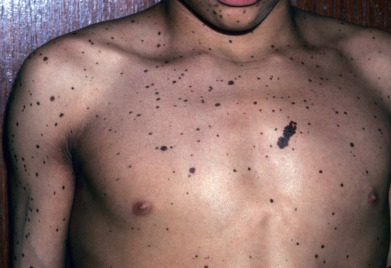
Patients with neurofibromatosis type I (NF-1, von Recklinghausen disease), which accounts for more than 85% of cases, typically present in infancy. NF-1 is either inherited autosomal dominantly or occurs due to de novo mutations. The most common initial manifestations are a large head circumference along with six or more café au lait macules. The typical café au lait macules of NF-1 should have a smooth margin and are oval or round and typically very clinically apparent ( Fig. 27.3 ). Most often, these macules are present at birth and almost always present by 1 year of age. The finding of six or more of these lesions measuring at least 1.5 cm in diameter is diagnostic, usually indicating NF-1. In children, the minimum diameter for a significant lesion is 0.5 cm. There are many children who have brown patches with jagged margins that appear splattered onto the skin or arranged in blaschkoid pattern and these are not typical for NF-1. Histologically, basilar hyperpigmentation is noted, and giant melanosomes may be seen. Axillary freckling ( Fig. 27.4 ) and/or inguinal freckling (Crowe sign) may also be present at birth and can extend to the neck and involve the inguinal, genital, and perineal areas.
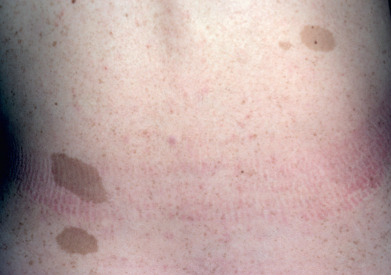
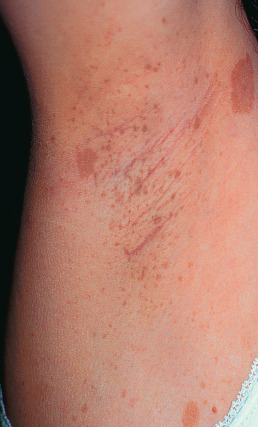
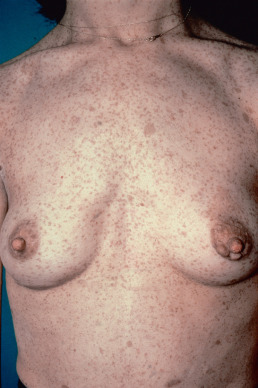
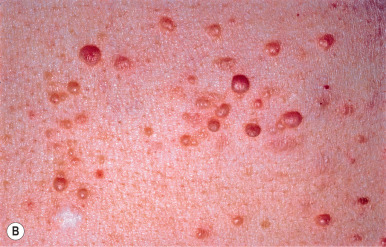
Neurofibromas ( Fig. 27.5 ) develop later in childhood and continue to develop into adulthood but are rarely present in infants unless there is a plexiform neurofibroma. Neurofibromas of the areolae occur in more than 90% of women with NF-1. Neurofibromas are soft tumors that can be pushed down into the panniculus by light pressure with the finger (“buttonholing”) and spring back when released. Histologically, these are well-circumscribed, but rarely encapsulated, spindle cell proliferations with an amphophilic myxoid stroma and many mast cells. The spindle cells have a wavy appearance. Neurofibromas result from proliferation of all supporting elements of the nerve fibers, including Schwann, perineurial, endoneurial, and mast cells and blood vessels. Axon stains demonstrate individual axons spread randomly throughout the tumor, in contrast to a schwannoma, where a nerve trunk is compressed at one edge of the tumor, but no axons are present within its bulk.
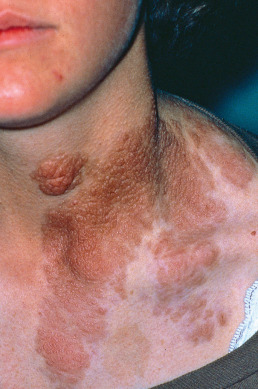
Subcutaneous plexiform neurofibromas are virtually pathognomonic of NF-1 and are often a manifestation of LOH. On palpation, these resemble a “bag of worms.” The overlying skin is usually hyperpigmented and may resemble a giant café au lait macule ( Fig. 27.6 ). Because the plexiform neurofibromas can take time to grow and manifest the larger café au lait may be the only marker of its presence in infancy. Plexiform neurofibromas within the spine can cause paralysis. Histologically, plexiform neurofibromas demonstrate numerous elongated encapsulated neurofibromas, often embedded in diffuse neurofibroma that involves the dermis and subcutaneous fat.
Many organ systems may be involved. Optic gliomas can be present in infancy and patients should be referred to an ophthalmologist immediately. Lisch nodules are found in the irides of about one quarter of patients under 6 years of age and in 94% of adult patients. Acromegaly, cretinism, hyperparathyroidism, myxedema, pheochromocytoma (<1%), or precocious puberty may be present. Patients with NF-1 can present with hypertension either due to renal artery stenosis or aortic coarctation (early in life) or a pheochromocytoma usually manifested later, and thus all patients with NF-1 should have their blood pressure closely monitored. Bone changes (usually erosive) may produce lordosis, kyphosis, and pseudoarthrosis, as well as spina bifida, dislocations, and atraumatic fractures. The pseudoarthrosis (pseudojoints) can be seen on imaging of long bones in childhood in some patients.
Patients with NF-1 are four times more likely to develop malignancies than the general population. Cutaneous neurofibromas rarely develop into malignant, peripheral nerve sheath tumors. A painful, growing or hardening lesion is an indication for biopsy. An increased incidence of breast carcinoma, Wilms tumor, rhabdomyosarcomas, gastrointestinal (GI) malignancies, and chronic myelogenous leukemia (CML) has also been reported. Children with NF-1 are 200–500 times more likely to develop malignant myeloid disorders than age-matched controls, and the risk for juvenile chronic myelomonocytic leukemia (JCMML) may be higher for those with juvenile xanthogranulomas (JXGs). Therefore some experts advocate for regular complete blood counts for the first few years in patients with NF-1 who have JXG.
Mental retardation, dementia, epilepsy, and a variety of intracranial malignancies may occur. Hypertelorism heralds a severe expression of neurofibromatosis with brain involvement. Diffuse interstitial lung disease occurs in 7% of patients.
Approximately 50% of cases of NF-1 represent new mutations. The gene for NF-1 codes for neurofibromin, a protein that negatively regulates signals transduced by Ras proteins. There is a high rate of spontaneous postzygotic mutation of this gene. Both alleles must be affected for the individual to grow a neurofibroma. In patients with the syndrome, there is germline loss of one allele, and each neurofibroma that develops represents a late spontaneous mutation knocking out the remaining allele.
Segmental neurofibromatosis (also called mosaic neurofibromatosis) arises from postzygotic somatic mutation ( Fig. 27.7 ). Early postzygotic mutation affecting the second allele in fetal life results in LOH affecting an entire Blaschko segment. Within this affected lesion any of the other features of NF-1 could occur, but in one series 29% of the patients were found to have features of NF-1 affecting other tissues and thus patients with segmental NF-1 should be monitored similarly to patients with nonsegmental NF-1. Diagnosis is by biopsying affected tissues for genetic testing because the peripheral blood may be negative.
Diagnosis
The diagnosis of NF-1 requires two or more of the following criteria to be fulfilled:
- 1.
Six or more café au lait macules with a greatest diameter of more than 5 mm in prepubertal individuals, and a greatest diameter of more than 15 mm in postpubertal individuals
- 2.
Two or more neurofibromas of any type or one plexiform neurofibroma
- 3.
Freckling in the axillary or inguinal regions
- 4.
Optic gliomas
- 5.
Two or more Lisch nodules
- 6.
Distinctive osseous lesion, such as a sphenoid dysplasia or thinning of the long-bone cortex with or without pseudarthrosis
- 7.
First-degree relative (parent, sibling, or offspring) with the disease
A diagnosis of NF-2 requires either of the following:
- 1.
Bilateral eighth cranial nerve masses, as demonstrated on computed tomography (CT) or magnetic resonance imaging (MRI)
- 2.
First-degree relative with NF-2 and either unilateral eighth nerve mass or two of the following: a neurofibroma, meningioma, glioma, schwannoma, and juvenile posterior subcapsular lenticular opacity
Although not listed in the previous criteria, the presence of nevus anemicus, xanthogranuloma, and glomus tumors is strongly associated with a diagnosis of NF-1, and the prevalence is high during the first 2 years of life, when other diagnostic criteria may be absent so they are vital to recognize. Nevus anemicus is usually found on the neck and upper chest, whereas the xanthogranulomas tend to be cephalic or genital.
Screening and Monitoring for Complications
In one study of 93 asymptomatic patients with NF-1 who underwent cerebral imaging, 12 optic gliomas were detected, suggesting that screening MRI or CT may be of value. Positron emission tomography (PET) scanning has shown some value in discriminating between benign and malignant tumors. The National Institutes of Health (NIH) consensus panel concluded that studies should be dictated by findings on clinical evaluation. It concluded that laboratory tests in asymptomatic patients are unlikely to be of value. In the majority of patients with NF-1, imaging studies should only be performed as indicated by signs or symptoms.
Therapy for individual symptomatic lesions is with surgical removal, but sirolimus has shown some efficacy for plexiform neurofibromas that are causing morbidity. Trials of targeted therapy to reduce the growth of cutaneous neurofibromas are ongoing and are likely to result in better treatment options for severely affected patients.
Type 2 neurofibromatosis (NF-2), central or acoustic neurofibromatosis, is distinguished by bilateral acoustic neuromas, usually in the absence of cutaneous lesions, although neurofibromas and schwannomas may occur. The gene for NF-2 encodes for merlin (schwannomin), a protein that links the actin cytoskeleton to cell surface glycoproteins and functions as a negative growth regulator. NF-2 patients, in contrast, often require imaging studies. Screening studies should include an audiogram and brainstem auditory evoked responses. MRI is the best imaging procedure for patients with evidence of hearing impairments or abnormal evoked responses. Tests of vestibular function may be useful, because eighth cranial nerve tumors develop on the vestibular division. A screening MRI should be performed by puberty. Other tests should be performed as dictated by signs and symptoms. Pediatric patients with NF-2 have a worse prognosis, with 75% demonstrating hearing loss, 83% visual impairment, and 25% abnormal ambulation. Type 3 (mixed) and type 4 (variant) forms resemble type 2 but have cutaneous neurofibromas. Patients with these types are at greater risk for developing optic gliomas, neurilemmomas, and meningiomas. These forms are inherited as autosomal dominant traits.
SPRED1 (Legius Syndrome)
Germline loss-of-function mutations in the SPRED1 gene have been associated with an NF-1–like phenotype with café au lait macules and axillary or inguinal freckling but no neurofibromas or lisch nodules (Legius syndrome). Patients present with similar café au laits and increased head circumference to NF-1, and most laboratories that do genetic testing for NF-1 will do SPRED1 reflexively if NF-1 testing is negative because the diseases match each other so closely. Developmental delay in patients with mosaic SPRED1 has now been reported as well.
Noonan Syndrome
Noonan syndrome is an autosomal dominant RASopathy associated with a webbed neck that mimics Turner syndrome but affects males and females equally. The other major features are a characteristic facies with hypertelorism, prominent ears, short stature, undescended testicles, low posterior neck hairline, cardiovascular abnormalities (e.g., pulmonary stenosis), and cubitus valgus. From 25%–40% of patients have dermatologic findings: lymphedema, short curly hair, dystrophic nails, tendency toward keloid formation, soft elastic skin, keratosis pilaris atrophicans (ulerythema of eyebrows), multiple granular cell tumors, and abnormal dermatoglyphics. Growth hormone can help patients achieve more normal stature.
Genetic changes in Noonan affect the RAS/MAPK pathway. The majority of Noonan cases are caused by defects in PTPN11. Patients with PTPN11 have more café au lait macules. Patients with Noonan syndrome may have SOS1 mutations associated with normal cognition and stature along with keratosis pilaris, RAF1 mutations entailing a high risk of hypertrophic cardiomyopathy. PTPN11 mutations predispose to juvenile myelomonocytic leukemia. When differentiating RAsopathies, feeding difficulties and developmental motor delay are the most common features with the cardiofaciocutaneous syndrome and Costello syndrome. Noonan-associated RASopathies appear to have an increased risk of lupus erythematosus.
Noonan Syndrome–Like Disorder With Loose Anagen Hair (NSLAH)
Recently a group of patients with Noonan-like features (short stature, typical facies, and pectus excavatum) along with loose anagen have been documented. They also have more hyperactive behavior, a hoarse voice, and more severe developmental delay. This is caused by a SHOC2 mutation (or more recently described with PPP1CB ). The hair demonstrates an anagen hair bulb (hockey stick or tube sock at the end) on dermoscopy, but recently trichorrhexis nodosa and trichoptilosis along with darkened or hairless skin have been described.
Noonan Syndrome With Lentigines (LEOPARD)
The LEOPARD syndrome—multiple lentigines, electrocardiographic conduction abnormalities, ocular hypertelorism, pulmonary stenosis, abnormal genitalia, retardation of growth, and sensorineural deafness—also known as multiple lentigines syndrome, cardiocutaneous syndrome, lentiginosis profusa syndrome, or progressive cardiomyopathic lentiginosis has been renamed Noonan syndrome with lentigines. The lentigines occur in nearly all patients and are small, dark-brown, polygonal, and irregularly shaped macules, usually measuring 2–5 mm in diameter that start around age 5 and increase dramatically over time. There are none in the mucosa. Individual lesions may be larger, even up to 1–1.5 cm. Melanoma has been described in these patients, so atypical lesions should be biopsied.
The LEOPARD syndrome shares many clinical features with Noonan syndrome. These are allelic disorders; patients with both syndromes ( Fig. 27.8 ) demonstrate mutations in the Noonan syndrome gene, PTPN11, although patients with BRAF, RAF1, and MAP2K1 mutations are described . Although the “R” in LEOPARD indicates growth retardation, some patients with the syndrome also exhibit mild mental retardation or speech difficulties. Many cases appear sporadically; however, inheritance as an autosomal dominant genetic trait has also been reported. Patients should be referred for cardiovascular and hearing evaluation and screened for cryptorchidism.
Costello Syndrome
Costello syndrome is characterized by growth retardation; failure to thrive in infancy; coarse facies; redundant velvety skin on the neck, palms, soles, and fingers; acanthosis nigricans; and nasal papillomata caused by HRAS mutations. Ventricular dilation is observed in more than 40% of cases. Hydrocephalus, brain atrophy, Chiari malformation, and syringomyelia may occur. Mild to moderate mental deficiency is frequently discovered, and most patients exhibit a characteristic sociable and friendly personality.
Cardiofaciocutaneous Syndrome
Cardiofaciocutaneous (CFC) syndrome is characterized by a distinctive facial appearance, heart defects, and mental retardation. Facial characteristics include high forehead with bitemporal constriction, downslanting palpebral fissures, hypoplastic supraorbital ridges, a depressed nasal bridge, and posteriorly angulated ears with prominent helices. The heart defects include pulmonic stenosis, atrial septal defect, and hypertrophic cardiomyopathy. Patients may have ectodermal abnormalities. The most frequent dermatologic findings in CFC patients involve the hair, which may be sparse, curly, fine or thick, or woolly or brittle. In the majority reported cases, the patient had dry, scaly, ichthyotic skin with follicular hyperkeratosis (keratosis pilaris, or keratosis pilaris atrophicans faciei). Other cutaneous findings include sparse or absent eyebrows and eyelashes, low posterior hairline, patchy alopecia, scant body hair, seborrheic dermatitis, eczema, lymphedema, hemangiomas, café au lait spots, pigmented nevi, hyperpigmented macules or stripes, cutis marmorata, and sacral dimples. Nail dystrophy, koilonychia, and dysplastic teeth have also been reported.
Most cases occur sporadically, but autosomal dominant transmission has been reported. Various subtypes relate to different genes in the RAS/MAPK pathway, including KRAS, BRAF, and MEK1 or MEK2 mutations.
Aoki Y, et al: Ras/MAPK syndromes and childhood hemato-oncological diseases. Int J Hematol 2013; 97: 30.
Bader-Meunier B, et al: Are RASopathies new monogenic predisposing conditions to the development of systemic lupus erythematosus? Semin Arthritis Rheum 2013; 43: 217.
Blakeley J: Development of drug treatments for neurofibromatosis type 2–associated vestibular schwannoma. Curr Opin Otolaryngol Head Neck Surg 2012; 20: 372.
Duan L, et al: Renal artery stenosis due to neurofibromatosis type 1. Eur J Med Res 2014; 19: 17.
Ferner RE, et al: Neurofibromatosis type 1 (NF1). Handb Clin Neurol 2013; 115: 939.
Ferrari F, et al: Juvenile xanthogranuloma and nevus anemicus in the diagnosis of neurofibromatosis type I. JAMA Dermatol 2014; 150: 42.
García-Romero MT, et al: Mosaic neurofibromatosis type 1. Pediatr Dermatol 2016; 33: 9.
Gelb, BD, Tartaglia M: Noonan syndrome with multiple lentigines. 2007 Nov 30 [Updated 2015 May 14]. I: Adam MP, Ardinger HH, Pagon RA, et al [Eds.] GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle.
Gripp KW, et al: A novel rasopathy caused by recurrent de novo missense mutations in PPP1CB closely resembles Noonan syndrome with loose anagen hair. Am J Med Genet A 2016; 170: 2237.
Gutmann DH, et al: Optimizing biologically targeted clinical trials for neurofibromatosis. Expert Opin Investig Drugs 2013; 22: 443.
Harrison B, Sammer D: Glomus tumors and neurofibromatosis. Plast Reconstr Surg Glob Open 2014; 2: e214.
Jobling RK, et al: Mosaicism for a SPRED1 deletion revealed in a patient with clinically suspected mosaic neurofibromatosis. Br J Dermatol 2017; 176: 1077.
Kane J, et al: Noonan syndrome with loose anagen hair associated with trichorrhexis nodosa and trichoptilosis. Clin Case Rep 2017; 5: 1152.
Madanikia SA, et al: Increased risk of breast cancer in women with NF1. Am J Med Genet 2012; 158A: 3056.
Martínez-Quintana E, et al: LEOPARD syndrome. Mol Syndromol 2012; 3: 145.
Morice-Picard F, et al: Cutaneous manifestations in Costello and cardiofaciocutaneous syndrome. Pediatr Dermatol 2013; 30: 665.
Myers A, et al: Perinatal features of the RASopathies. Am J Med Genet A 2014; 164: 2814.
Niemeyer CM: RAS diseases in children. Haematologica 2014; 99: 1653.
Pasmant E, et al: Neurofibromatosis type 1. J Med Genet 2012; 49: 483.
Pećina-Šlaus N: Merlin, the NF2 gene product. Pathol Oncol Res 2013; 19: 365.
Rauen KA: The RASopathies. Annu Rev Genomics Hum Genet 2013; 14: 355.
Treglia G, et al: Usefulness of whole-body fluorine-18-fluorodeoxyglucose positron emission tomography in patients with neurofibromatosis type 1. Radiol Res Pract 2012; 2012: 431029.
Tumurkhuu M, et al: A novel SOS1 mutation in Costello/CFC syndrome affects signaling in both RAS and PI3K pathways. J Recept Signal Transduct Res 2013; 33: 124.
Vranceanu AM, et al: Quality of life among adult patients with neurofibromatosis 1, neurofibromatosis 2 and schwannomatosis. J Neurooncol 2013; 114: 257.
Weiss B, et al: Sirolimus for progressive neurofibromatosis type 1–associated plexiform neurofibromas. Neuro Oncol 2015; 17: 596.
Epidermal Nevus Syndromes
Epidermal nevi are overgrowths of specific epidermal structures and can be isolated to skin findings or if more widespread or extensive can be a marker for a syndrome. Important clues to the diagnosis of specific epidermal nevus (EN) syndromes include linear lesions following blaschkoid lines because the lesions and syndromes are caused by postzygotic mutations leading to mosaicism. Many of the following syndromes are reviewed in other chapters: linear nevus sebaceous (NS) in Schimmelpenning syndrome, NS and papular nevus spilus in phacomatosis pigmentokeratotica, soft white hair in angora hair nevus syndrome (Schauder syndrome), breast hypoplasia in Becker nevus syndrome, mosaic R248 C mutation in fibroblast growth factor receptor 3 EN syndrome (characterized by soft velvety EN and CNS abnormalities), and acral strawberry papillomatous lesions on tips of fingers or toes in CHILD syndrome (see Congenital Hemidysplasia with Ichthyosiform Erythroderma and Limb Defects [CHILD] Syndrome , later in this chapter). Other EN syndromes include nevus trichilemmocysticus (cysts in blaschkoid distribution), didymosis aplasticosebacea (NS with aplasia cutis congenita), SCALP syndrome (NS, CNS malformations, aplasia cutis, limbal dermoid, and pigmented nevus), Gorbello syndrome (systematized linear velvety EN with bone defects), NEVADA syndrome (nevus epidermicus verrucosus with angiodysplasia and aneurysms), and CLOVE syndrome (congenital lipomatous overgrowth [see Chapter 28 ], vascular malformations, and EN with nonprogressive proportionate overgrowth).
Cirstea IC, et al: Diverging gain-of-function mechanisms of two novel KRAS mutations associated with Noonan and cardio-facio-cutaneous syndromes. Hum Mol Genet 2013; 22: 262.
Hafner C, et al: Keratinocytic epidermal nevi are associated with mosaic RAS mutations. J Med Genet 2012; 49: 249.
Hafner C, et al: Mosaic RASopathies. Cell Cycle 2013; 12: 43.
Laura FS: Epidermal nevus syndrome. Handb Clin Neurol 2013; 111: 349.
Proteus Syndrome
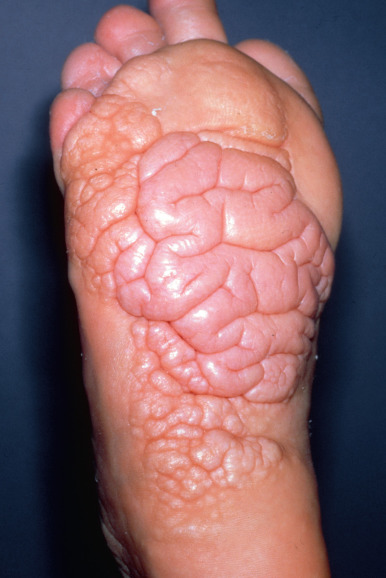
Proteus syndrome is an overgrowth syndrome caused by an AKT1 mutation. The mutation appears to be mosaic and activating leading to overgrowth. Proteus shares many features with other syndromes such as PTEN hamartoma syndrome and CLOVE syndrome (see Chapter 28 ) because they are within a common cellular pathway. Proteus syndrome is named after the Greek god Proteus. The syndrome has protean manifestations that include disproportionate, asymmetric, and distorting segmental overgrowth; cerebriform plantar hyperplasia ( Fig. 27.13 ); epidermal nevi; patchy dermal hypoplasia; macrocephaly; hyperostosis; muscular hypoplasia; hypertrophy of long bones; vascular malformations of the capillary, venous, or lymphatic types; lipomas, lipohypoplasia; fatty overgrowth; bullous lung alterations; intellectual disability; seizures; brain malformations; and deep vein thrombosis. The cerebriform hyperplasia is histopathologically a connective tissue nevus made of collagen and can occur most commonly on the plantar foot but also the palm, abdomen, and nose and is a major diagnostic criterion of Proteus syndrome and very suggestive if present. The vascular lesions are capillary malformations and tend to be darker purple and geometric. Patients with a greater number of cutaneous lesions also have the most extracutaneous abnormalities. There is a high risk of deep vein thrombosis. The vascular stains and epidermal nevi may be the first manifestations because the overgrowth can take time to present (different from CLOVE syndrome) that has similar features. There is progressive severe bony overgrowth in Proteus syndrome that can be extremely distorting.
Cohen MM Jr: Proteus syndrome review. Clin Genet 2014; 85: 111.
Lindhurst MJ, et al: AKT1 gene mutation levels are correlated with the type of dermatologic lesions in patients with Proteus syndrome. J Invest Dermatol 2014; 134: 543.
Rozas-Muñoz E, et al: Vascular stains: proposal for a clinical classification to improve diagnosis and management. Pediatr Dermatol 2016; 33: 570.
Phakomatoses
The phakomatoses are the various inherited disorders of the CNS associated with cutaneous and often ocular involvement. The term was more helpful in organizing genodermatoses before the current genetic pathophysiologic classifications. Phakomatoses include tuberous sclerosis, neurofibromatosis, von Hippel–Lindau disease, ataxia-telangiectasia, nevoid basal cell carcinoma syndrome, nevus sebaceous, and Sturge-Weber syndrome. Many of these disorders have been grouped into more specific groups in this book due to more specific understanding of the pathogenisis of each syndrome.
Tuberous Sclerosis
Tuberous sclerosis was first described by Desiree-Magloire Bourneville in 1880. The classic triad of adenoma sebaceum ( Fig. 27.9 ), mental deficiency, and epilepsy, however, is present in only a minority of patients. Other associated features include periungual fibromas, shagreen plaques (collagenoma), oral papillomatosis, gingival hyperplasia, ash-leaf hypomelanotic macules, skin fibromas, and café au lait spots.
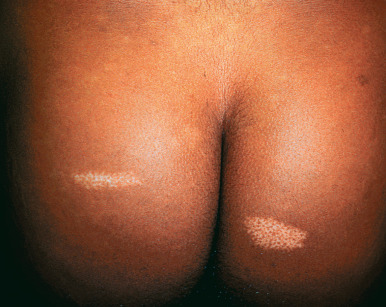
The first cutaneous lesions are typically the white patches that are oval and shaped like an “ash leaf” ( Fig. 27.10 ). These congenital white, leaf-shaped macules, also called hypomelanotic macules, are found in 85% of patients with tuberous sclerosis and range in number from 1 to 100, although 3 lesions greater than 5 mm must be present to satisfy the criteria. Occasional patients may not develop the macules until 6–8 years of age. These may be shaped like an ash leaf, but linear and confetti-type white macules may also be present. Wood’s light examination will highlight these lesions because they can be subtle. Focal poliosis (localized tufts of white hair) may be present at birth. Solitary ash-leaf macules can occur in the general population and may be difficult to distinguish from pigmentary mosaicism and nevus depigmentosus.
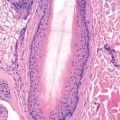
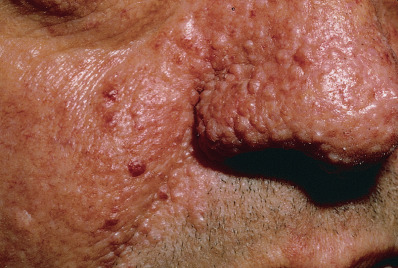
Adenoma sebaceum lesions (angiofibromas) are 1–3 mm, yellowish red, translucent, discrete, waxy papules that are distributed symmetrically, principally over the cheeks, nose, and forehead. These lesions are present in 90% of patients older than 4 years, persist indefinitely, and may increase in number. A fibrous forehead plaque (newly named “fibrous cephalic plaque” because they can be anywhere on the head) is usually on the forehead and histopathologically appears to be an angiofibroma. Angiofibromas have also been reported in patients with multiple endocrine neoplasia (MEN-1) and the Birt-Hogg-Dube syndrome.
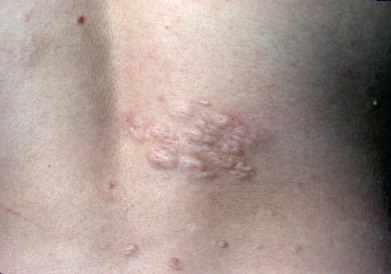
Shagreen plaque is named after a type of leather tanned to produce knobs on the surface, resembling shark skin. Patches of this type of “knobby” skin, varying from 1–8 cm in diameter, are found on the trunk, most often on the lumbosacral area ( Fig. 27.11 ). These are connective tissue nevi composed almost exclusively of collagen, occur in 40% of patients, and develop in the first decade of life.
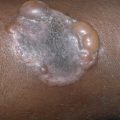
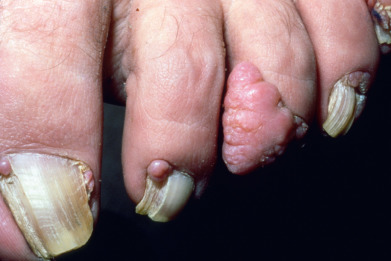
Koenen tumors (periungual angiofibromas) occur in 50% of patients ( Fig. 27.12 ). The tumors are small, digitate, protruding, asymptomatic, and periungual/subungual. They appear at puberty. Similar lesions may occur on the gingiva. Nails may also demonstrate longitudinal grooves, long leukonychia, and short red streaks.
Gingival fibromas in the mouth can occur on the buccal mucosa, the labial mucosa, and the tongue.
Mental deficiency, usually appreciated early in life, is present in 40%–60% of patients, varying widely in its manifestations. Epilepsy also occurs, is variable in its severity, and usually also presents early in life. Between 80% and 90% of patients have seizures or nonspecific electroencephalographic abnormalities. Hamartomatous proliferations of glial and neuronal tissue produce potato-like nodules in the cortex. X-ray evaluation will reveal these once calcified, but CT, cranial ultrasonography, and MRI may define these lesions as early as 6 weeks of age and thus are useful in making an early diagnosis. These brain tumors may progress to gliomas. Subependymal nodules (“candle drippings”) are similar lesions in the ventricular walls. Astrocytomas may also occur. Forehead plaques may be a marker for more serious intracranial involvement.
Retinal tumors (phakomas) are optic nerve or retinal nerve hamartomas. Various ophthalmologic findings, such as pigmentary changes, nystagmus, and angioid streaks, occur in 50% of patients. Renal hamartomas (angiomyolipomas, cystic disease, fibroadenomas, or mixed tumors) and cardiac tumors (rhabdomyomas in 43%) may also occur. Renal angiomyolipomas are bilateral and cause renal failure. The cardiac rhabdomyomas may only be seen prenatally and regress after birth but are highly specific for tuberous sclerosis (TS). Pulmonary lymphangioleiomyomatosis is more common in women, especially in their thirties and forties, and can lead to progressive respiratory failure or spontaneous pneumothorax. The condition is characterized by diffuse proliferation of smooth muscle cells and cystic degeneration of the pulmonary parenchyma, associated with the perivascular epithelioid cells (“PEC” cells) implicated in various PEComas. Almost half of patients with TS have bony abnormalities such as bone cysts and sclerosis, which can be seen on x-ray evaluation. Five or more pits in the enamel of permanent teeth are a marker for this disease.
Tuberous sclerosis is a common inherited autosomal dominant disease with highly variable penetrance. Prevalence estimates range from 1 in 5800 to 1 in 15,000. Up to 50% of cases may result from spontaneous mutations. There are two genes, the mutations of which produce indistinguishable phenotypes TSC1 and TSC2. These are tumor suppressor genes. TSC2 encodes for tuberin, a putative guanosine triphosphatase (GTPase)–activating protein for rap1 and rab5. TSC1 encodes for hamartin, a novel protein with no significant homology to tuberin or any other vertebrate protein. Hamartin and tuberin associate physically in vivo, suggesting that they function in the same complex rather than in separate pathways. This interaction of tuberin and hamartin explains the indistinguishable phenotypes caused by mutations in either gene. Hamartomas frequently demonstrate loss of the remaining normal allele (loss of heterozygosity).
Diagnosis
The major criteria for TS are three or more hypomelanotic macules, three or more angiofibromas or a fibrous cephalic plaque, two or more periungual fibromas, shagreen patch, retinal hamartomas, cortical dysplasia, subependymal nodules, subependymal giant cell astrocytoma, cardiac rhabdomyoma, lymphangioleiomyomatosis, and angiomyolipomas.
The minor criteria are confetti-like macules, three or more dental enamel pits, two more intraoral fibromas, retinal achromic patch, multiple renal cysts, and nonrenal hamartomas.
A diagnosis of TS is made either with positive TSC 1 or 2 testing or with two major or one major and two or more minor criteria. A possible diagnosis is with one major or two or more minor criteria.
Workup should include a full skin examination for cutaneous features including a Wood’s light examination. If x-ray examination fails to show calcified intracranial nodules, ultrasonography, CT, or MRI should be performed. Funduscopic examination, hand and foot x-ray evaluation, and renal ultrasonography are often revealing in a patient with few clinical findings; up to 31% of asymptomatic parents have been identified using these tests.
Treatment
Adenoma sebaceum can be treated by shaving, dermabrasion, or laser therapy, but topical rapamycin ranging from 0.1%–1% applied to the lesions can eradicate them noninvasively. Lesions are likely to recur, requiring maintenance treatment. Everolimus was the first mTOR inhibitor approved in the United States and Europe as a treatment for subependymal giant cell astrocytomas. Clinical evidence also supports the use of mTOR inhibitors, including sirolimus, in a variety of tuberous sclerosis complex–associated disease manifestations, including facial angiofibromas, renal angiomyolipoma, and epilepsy. Cranial irradiation of astrocytomas should be avoided because this may result in the subsequent development of glioblastomas.
Cudzilo CJ, et al: Lymphangioleiomyomatosis screening in women with tuberous sclerosis. Chest 2013; 144: 578.
Curatolo P, et al: mTOR inhibitors in tuberous sclerosis complex. Curr Neuropharmacol 2012; 10: 404.
Ebrahimi-Fakhari D, et al: Topical rapamycin for facial angiofibromas in a child with tuberous sclerosis complex (TSC). Dermatol Ther (Heidelb) 2017; 7: 175.
Liebman JJ, et al: Koenen tumors in tuberous sclerosis. Ann Plast Surg 2014; 73: 721.
Northrup H, et al: Tuberous sclerosis complex diagnostic criteria update. Pediatr Neurol 2013; 49: 243.
Teng JM, et al: Dermatologic and dental aspects of the 2012 International Tuberous Sclerosis Complex Consensus Statements. JAMA Dermatol 2014; 150: 1095.
Tu J, et al: Topical rapamycin for angiofibromas in paediatric patients with tuberous sclerosis. Australas J Dermatol 2014; 55: 63.
Von Hippel–Lindau Syndrome
Von Hippel–Lindau syndrome is an autosomal dominant disorder consisting of retinal angiomas, cerebellar medullary angioblastic tumors, pancreatic cysts, and renal tumors and cysts. Ocular lesions may lead to retinal detachment. Pheochromocytoma has been associated in several kindreds with von Hippel–Lindau disease. Usually, the skin is not involved, although capillary malformations on the head and neck and café au lait macules have been described.
Patiroglu T, et al: Cerebellar hemangioblastoma associated with diffuse neonatal hemangiomatosis in an infant. Childs Nerv Syst 2012; 28: 1801.
Ataxia-Telangiectasia
Also known as Louis-Bar syndrome, ataxia-telangiectasia (AT) consists of cerebellar ataxia, oculocutaneous telangiectasia, and sinopulmonary infections. Patients may have a marked IgA deficiency, with decreased lymphocytes and a small to absent thymus. In 80% of cases, IgA is absent or deficient; in 75%, absent or deficient IgE is seen; and in 50%, IgG is very low. There also can be T-cell lymphopenia. AT is caused by a mutation in the ATM gene. It is usually first noted when the child begins to walk. There is awkwardness and a swaying gait, which results in the child needing to use a wheelchair by about 10 years of age. Choreic and athetoid movements and pseudopalsy of the eyes are other features. Fine telangiectases appear on the exposed surfaces of the conjunctiva at about age 3. Telangiectases also appear later on the butterfly area of the face, inside the helix and over the backs of the ears, in the roof of the mouth, in the necklace area, in the flexures, and over the dorsa of the hands and feet. The skin tends to be dry and coarse and over time becomes tight and inelastic, as in scleroderma. Atrophic, granulomatous, scarring plaques may occur. Granulomatous plaques occur in various immunodeficiency syndromes including cartilage hair hypoplasia, combined variable immunodeficiency and DNA ligase deficiency. Rubella virus has been documented by PCR in the granulomatous lesions and likely is a result of the immune system not being able to handle the killed virus that is introduced during vaccination that occurs with the live vaccine at age 1 before knowing the child has immunodeficiency.
Other cutaneous stigmata are café au lait patches, hypopigmented macules, melanocytic nevi, hypertrichosis, seborrheic dermatitis, premature graying and sparsity of the hair, and progeroid features. The most common types of malignancy are lymphomas, usually of the B-cell type, and leukemias. It has been shown that homozygous patients also have a higher risk of breast cancer—100 times higher than age-matched controls. Heterozygous carriers share the defective repair of radiation-induced damage, and there is a threefold to fivefold higher risk for development of neoplasms, especially breast cancer, in heterozygotes under age 45. The ovaries and testicles do not develop normally. There is deficient thymus development, with absence of Hassall’s corpuscles and a lack of T-helper cells. Suppressor T cells are normal.
Early death from bronchiectasis occurs in more than half these patients, most of whom have recurrent sinus and lung infections that begin between 3 and 8 years of age.
Ataxia-telangiectasia is transmitted as an autosomal recessive trait, and heterozygotes, although they lack clinical findings, are cancer prone. The ATM gene is involved in cell cycle control, meiotic recombination, telomere length monitoring, and DNA damage response. Affected cells are hypersensitive to ionizing radiation.
Early diagnosis can be difficult and the most frequent misdiagnosis is cerebral palsy. The ataxia, along with telangiectasias, persistently elevated levels of α-fetoprotein, and carcinoembryonic antigen, along with the immunodeficiency, are useful in early diagnosis. Genetic testing is diagnostic.
Greenberger S, et al: Dermatologic manifestations of ataxia-telangiectasia syndrome. J Am Acad Dermatol 2013; 68: 932.
Knoch J, et al: Rare hereditary diseases with defects in DNA-repair. Eur J Dermatol 2012; 22: 443.
Rothblum-Oviatt C et al: Ataxia telangiectasia. Orphanet J Rare Dis 2016; 11: 159.
Zakko L, et al: Von Hippel–Lindau syndrome. In Wu G, Selsky N, Grant-Kels J [Eds.] Atlas of Dermatological Manifestations of Gastrointestinal Disease. New York, NY: Springer, 2013.
Epidermosysis Bullosa
Epidermolysis bullosa (EB) is a group of rare genetic disorders characterized by skin fragility with formation of blisters from minor physical injury. EB occurs due to congenital absence of proteins involved in keeping the epidermis together or connected to the dermis. Autoimmune blistering diseases often target similar proteins but are acquired. EB has been reclassified based on where the splitting of the skin occurs. The severity of EB subtypes is associated with the location of blistering, extent of loss of the skin, and other abnormalities associated with the protein that is defective or absent. The split in EB simplex (EBS) is within the epidermis (and is further divided into suprabasal and basal), junctional EB (JEB) splits within the basement membrane, and dystrophic EB (DEB) splits below the basement membrane. There are mixed pattern as well (Kindler). The inherited types of EB are classified as listed in Box 27.1 .
Intraepidermal
- •
Epidermolysis bullosa (EB) simplex, generalized intermediate, KRT5 or KRT14 mutation
- •
EB simplex, localized, KRT5 or KRT14 mutation
- •
EB, generalized severe, KRT5 or KRT14 mutation
- •
EBS with muscular dystrophy, pyloric atresia and Ogna are caused by Plectin mutations
- •
EB simplex-migratory circinate is caused by keratin 5 mutations
- •
EB simplex with mottled pigmentation, KRT5 mutation
- •
EBS with muscular dystrophy, pyloric atresia and Ogna (above) are caused by Plectin mutations)
- •
EB superficialis
- •
Acantholytic EB simplex, DSP or JUP mutations
- •
Acral peeling skin syndrome is caused by TGM5 mutation
- •
Skin fragility syndromes
- •
Skin fragility–wooly hair syndrome (desmoplakin mutation)
- •
Skin fragility (plakoglobin mutation)
- •
Skin fragility–ectodermal dysplasia syndrome (plakophilin mutation)
- •
- •
EB simplex autosomal recessive–BP230 mutation
- •
EB simplex autosomal recessive–exophilin 5 mutation
- •
EB simplex autosomal recessive– K14 mutation
Junctional (Intralamina Lucida)
- •
Junctional epidermolysis bullosa (JEB), generalized severe, LAMA3, LAMB3, or LAMC2 mutations
- •
JEB, generalized intermediate, LAMA3, LAMB3, LAMC2, or COL17A1 mutations
- •
JEB localized
- •
JEB with pyloric atresia (α6β4-integrin)
- •
JEB, late onset (Collagen XVII mutation)
- •
JEB with respiratory and renal involvement (α3-integrin subunit)
- •
JEB inversa
Dermolytic or Dystrophic (Sublamina Densa)
Dominant Forms
- •
Dystrophic EB, generalized, COL7A1 mutation
- •
EB pruriginosa
- •
Pretibial EB
- •
Bullous dermolysis of the newborn
Recessive Forms
- •
Generalized severe, collagen VII absent, COL7A1 mutations
- •
Generalized intermediate, COL7A1 mutations
- •
Bullous dermolysis of the newborn, COL7A1 mutations
- •
Localized (various types, including EB pruriginosa and pretibial EB)
Some of the mutations leading to EB are in genes that expressed in other tissues and therefore have extracutaneous manifestations. Esophageal and laryngeal complications are seen primarily in recessive dystrophic EB but may be present in JEB (Herlitz). Pyloric atresia can occur in JEB. Ocular lesions may be severe in dystrophic EB, and mild lesions have been reported in simplex and junctional disease. Congenital absence of the leg skin (Bart syndrome) presents with geometric loss of large areas of skin often on the medial legs extending to the calves possibly from one leg rubbing on the other in utero. This phenotype can be seen in multiple different forms of EB although it is most commonly reported with dystrophic EB.
Clinical findings and routine histologic features overlap in EB. Electron microscopy (EM) studies and immunofluorescent mapping can be used to determine the location of the split and the presence or absence of certain proteins. Immunofluorescent mapping may define the level of the split without resorting to EM. By staining biopsy specimens for normal components of the basement membrane zone (BMZ), such as bullous pemphigoid antigen, laminin, type IV collagen, or LDA-1 antigen, the level of the split may be determined by whether the antigen localizes at the roof or base of the blister. Biopsy should be done by inducing a new blister (usually with a pencil eraser) in clinically normal skin and sending to a specialized laboratory. EM and immunofluorescence do not always fully differentiate between various phenotypes that can be caused by mutations in the same genes, and genetic testing can be done to confirm a diagnosis. Now that genetic panels are available that include nearly all described EB mutations and are less expensive and faster than previously, it is reasonable to start with genetic testing because it will avoid a procedure and give a more definitive diagnosis. However, caution is advised predicting severity from genetic testing because not all patients with the same mutation will have the same clinical symptoms.
Intraepidermal Forms (Epidermolysis Bullosa Simplex)
Suprabasal Epidermolysis Bullosa Simplex
Suprabasal forms of EBS are caused by defects in transglutaminase 5, plakophilin, desmoplakin, and plakoglobin. They present with skin fragility and peeling.
Acral peeling skin syndrome is caused by transglutaminase 5 mutations and leads to superficial peeling, especially on the palms and soles, and heals without scarring.
EBS superficialis presents with superficial blisters and has no causal known mutation.
Skin fragility syndromes include EBS desmoplakin skin fragility and woolly hair syndrome, skin fragility–ectodermal dysplasia syndrome, and skin fragility–plakoglobin deficiency. Mutations in the genes that encode the plakin family (desmoplakin, plakoglobin, and plakophilin) can present with widely variable phenotypes, including with systemic findings.
Skin Fragility (Plakoglobin Mutation)
Plakoglobin mutations can affect skin, hair and heart. Different mutations can lead to isolated skin findings of fragility or isolated cardiac findings. The phenotype of wooly hair, palmoplantar keratoderma and cardiomyopathy is called Naxos disease.
Skin Fragility and Woolly Hair Syndrome (Desmoplakin Mutation)
Mutations in desmoplakin can lead to multiple different phenotypes depending in part on the specific mutation because desmoplakin is expressed in skin, hair, nails, and cardiac tissue. Skin fragility with wooly hair is isolated to the skin. Desmoplakin mutations can also cause a striate PPK in isolation, a striate PPK with wooly hair and cardiomyopathy (Carvajal syndrome), cardiac disease in isolation, or a lethal acantholytic EB with complete alopecia.
Skin Fragility–Ectodermal Dysplasia Syndrome (Plakophilin Mutation)
This syndrome is characterized by inflammation around the mouth with fissuring, nail dystrophy, trauma-induced skin fragility, and defects of the hair, nails, and sweat glands. Trauma-induced blisters are generalized with skin tearing noted on the pressure points, especially after prolonged standing or walking. Plakophilins are not expressed in cardiac tissue so there are no cardiac findings thus far reported.
Epidermolysis Bullosa Simplex, Basalar
There are multiple forms of EBS caused by mutations in the genes encoding keratins 5 or 14, and the severity and pattern depend on the how much the mutation disrupts function. Basal EBS mutations in Krt5 or Krt14 can cause EBS localized, EBS generalized-severe, or EBS generalized-intermediate. Mutations in Krt5 cause EBS with mottled pigmentation and EBS migratory circinate.
Mutations in K14 can cause an autosomal recessive EBS (EBS autosomal recessive-K14). EBS with muscular dystrophy, pyloric atresia, and Ogna are caused by plectin mutations. Two other types of autosomal recessive EBS are caused by BPAG-1 or exophillin 5 mutations.
Epidermolysis Bullosa Simplex, Localized
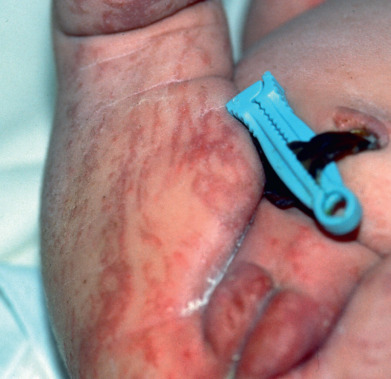
Recurrent bullous eruption of the hands and feet is autosomal dominantly determined and appears in a chronic form in early childhood with exacerbations as an adult based on activities that increase friction. The Weber-Cockayne designation has been dropped in recent classification schemes. The lesions worsen during hot weather and when the patient is subjected to prolonged walking or marching, as in military service. Hyperhidrosis may be an associated finding. In localized EBS, the bullae are intraepidermal and suprabasal, and healing occurs without scarring.
Application of aluminum chloride hexahydrate in anhydrous ethanol (Drysol) on the normal skin of hands and feet twice a day has been shown to reduce blistering in this form of EBS. After 2 weeks of daily therapy, the patient can be switched to weekly or twice-weekly applications.
Epidermolysis Bullosa Simplex, Generalized Intermediate
The generalized type of EBS, dominantly inherited with complete penetrance, occurs in 1 in 500,000 births. It is characterized by the development of vesicles, bullae, and sometimes milia over the joints of the hands, elbows, knees, and feet, as well as other sites subject to repeated trauma. The child is affected at birth or shortly thereafter, with improvement within the first few months, but there are more blisters when the child begins crawling. The blistering is worse during the summer and improves during the winter. The lesions are sparse and do not lead to severe atrophy. Usually, the mucous membranes and nails are not involved. EBS is usually milder than other forms of EB.
Inherited as an autosomal dominant trait, EBS is a disease in which keratin gene mutations cause the production of defective intermediate filaments, that lead to epidermal basal cell fragility and subsequent blistering. The genes encoding keratins 5 or 14 (expressed in the basal layer) are mutated. Patients heterozygous for abnormal keratin 14 have blistering limited to the hands and feet, but homozygotes have more severe and widespread blistering of the skin and mucous membranes. Separation occurs through the basal cell layer.
Epidermolysis Bullosa Simplex, Generalized Severe
In this autosomal dominant variant of EBS, active blisters ( Fig. 27.14 ) with circinate configuration occur in infancy. Milia may develop, but there is no scarring. The oral mucosa is involved. Nails are shed but may regrow, sometimes with dystrophy. Blistering lessens with age. Hyperkeratosis of the palms and soles may occur. Histologically, the split is through the basal layer, and tonofilaments are clumped on EM. Point mutations have been shown in keratin 5 and 14 genes.
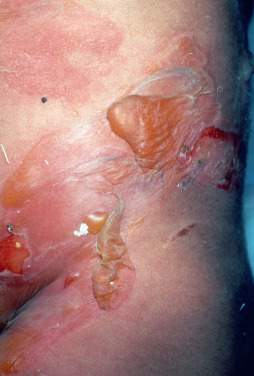
Epidermolysis Bullosa Simplex With Mottled Pigmentation
Patients have been reported with autosomal dominant EBS with congenital scattered hyperpigmented and hypopigmented macules that fade slowly after birth. The remaining features are similar to those of generalized EBS. Ultrastructural studies show vacuolization of the basal cell layer.
Epidermolysis Bullosa Simplex Migratory Circinate
Affected individuals present with slowly advancing erythema with blisters, and the lesions resolve with pigmentation. The mutation in EBS migratory circinate is the same as mottled pigmentation, so they are likely on a spectrum with some cases showing more erythema in early life and mottled pigmentation later.
Epidermolysis Bullosa Simplex With Muscular Dystrophy
A form of EBS is associated with late-onset neuromuscular disease and is caused by plectin mutation. Widespread blistering at birth is associated with scarring, milia, atrophy, nail dystrophy, dental anomalies, laryngeal webs, and urethral strictures. Progressive muscular dystrophy with weakness and wasting begins in childhood or later so patients should be followed over time.
Epidermolysis Bullosa Simplex With Pyloric Atresia
EBS with pyloric atresia is also caused by plectin mutation as well as integrin beta-4 and alpha-6 because integrin beta-4 binds plectin in the hemidesmosome. Late urologic complications have been described in patients with EBS with pyloric atresia.
Epidermolysis Bullosa Simplex (Ogna)
Generalized bruising and hemorrhagic blisters occur. EBS is transmitted as an autosomal dominant trait. At birth there are small, acral, traumatic sanguineous blisters. The basal keratinocytes in this syndrome do not stain with antiplectin antibodies.
Junctional Forms
Junctional epidermolysis bullosa (JEB) presents with severe generalized blistering usually at birth ( Fig. 27.15 ), and in most forms extensive denudation may prove fatal. JEB is caused by mutations in three genes: LAMA3, LAMB3, or LAMC2, that code for polypeptide subunits of laminin 332 (also referred to as laminin 5), COL XVII, and integrins alpha-3 or -6 or beta-4.
Junctional Epidermolysis Bullosa
JEB can classified as localized, generalized, or generalized severe. The more severe phenotypes are characterized by more blisters, scarring, and intraoral involvement. There is little to no GI, ocular genitourinary, or respiratory involvement in the less severe forms. JEB is characteristic perioral and perinasal hypertrophic granulation tissue. Eventually, the lesions heal without scarring or milia formation, but erosions may persist for years. Dysplastic teeth are common. Laryngeal and bronchial lesions may cause respiratory distress and death in more severe forms. Additional systemic complications include GI tract, gallbladder, corneal, and vaginal disease. Patients who survive infancy have growth retardation and, often, moderate to severe refractory anemia.
In addition to good wound care and control of infection, epidermal autographs of cultured keratinocytes, isolated from clinically uninvolved skin and grown on collagen sponges, may be useful for chronic facial erosions.
Junctional Epidermolysis Bullosa With Pyloric Atresia
This rare autosomal recessive inherited form of JEB presents at birth with severe mucocutaneous fragility and gastric outlet obstruction. Often the ears are crumpled and misshapen. Even if the pyloric atresia is repaired, the neonates may die of the severity of their skin disease. If they survive the neonatal period, the blistering diminishes. Persistent scarring of the urinary tract may occur, however, with stenosis of the ureteral-vesicular junction, requiring numerous urologic procedures. This syndrome is usually caused by a genetic mutation in either the α6- or β4-integrin genes ( ITGA6 and ITGB4 ). This α6β4-integrin complex is uniquely expressed on epithelial surfaces.
Other Forms of Junctional Epidermolysis Bullosa
JEB with late onset is caused by collagen XVII mutations and is generally more mild than typical JEB. JEB with respiratory or renal involvement is caused by mutations in integrin alpha-3.
Dermolytic or Dystrophic Forms of Epidermolysis Bullosa
The cause of dystrophic EB in both autosomal dominant and autosomal recessive inherited forms is mutation in the COL7A1 gene encoding for type VII collagen. The anchoring fibrils in these patients are defective or deficient. Presumably, because of antigen exposure, anti–type VII collagen, anti-BP180, and anti-BP230 autoantibodies may be detected.
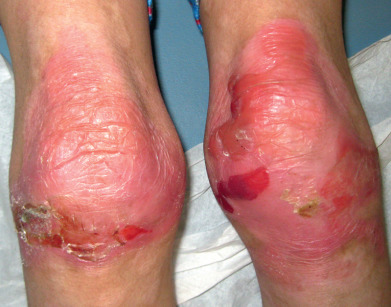
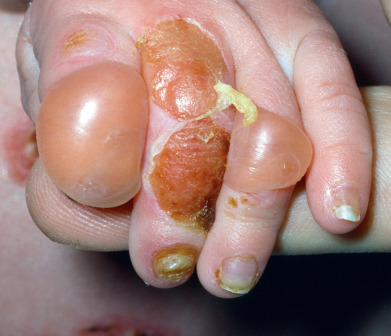
Dominant Dystrophic Epidermolysis Bullosa
Dominant dystrophic EB (DDEB) typically presents at or soon after birth with blisters but the overall course of it is favorable. The blisters are most pronounced over the joints, especially over the toes, fingers, knuckles, ankles, and elbows ( Figs. 27.16 and Fig. 27.17 ). Spontaneous, flesh-colored, scarlike (albopapuloid) lesions may appear on the trunk, often in adolescence, with no previous trauma and may be associated with more severe disease. The nails may be thickened. Usually, Nikolsky sign is present, and frequently the accumulated fluid in a bulla can be moved under the skin several centimeters away from the original site. Healing usually occurs with scarring and atrophy. Milia are often present on the rims of the ears, dorsal surfaces of the hands, and extensor surfaces of the arms and legs.