Fronto-Orbital Distraction
Jesse A. Taylor
Wen Xu
DEFINITION
Craniosynostosis affects 1 in 2500 live births worldwide and is defined as the premature fusion of one or more cranial sutures.
Craniosynostosis may affect a single suture or multiple sutures and may be idiopathic or associated with an underlying syndrome.
The most frequently affected suture is the sagittal suture, followed by the coronal suture, the metopic suture, and the lambdoid suture.
Cranial vault distraction allows for gradual expansion of bone and soft tissues of the cranium without leaving behind any indwelling hardware in the growing craniofacial skeleton.
ANATOMY
There are four major calvarial sutures (metopic, sagittal, coronal, and lambdoid) that are patent at birth but close in a predictable and sequential manner in the first few years of life.
The metopic suture divides the two halves of the frontal bone in the sagittal plane.
The sagittal suture is located along the midline and divides the left and right parietal bones.
The coronal suture is located between the frontal bone and the parietal bone and can be divided into a left coronal suture and a right coronal suture.
The lambdoid suture is located between the parietal bone and the occipital bone and is continuous inferiorly with the occipitomastoid suture.
The anterior fontanelle is a diamond-shaped membranefilled space located at the junction of the metopic, sagittal, and coronal sutures that is present in infants until approximately 18 months of age.
The posterior fontanelle is a triangle-shaped space located at the junction of the sagittal and lambdoid sutures that ossifies 2 to 3 months after birth.
PATHOGENESIS
Craniosynostosis involving a single suture is typically an isolated defect with no known direct cause and is not associated with a syndrome.
Presence of in utero fetal head constraint is associated with an increased incidence of craniosynostosis.1
Other risk factors include endocrine disturbances (hyperthyroidism, mucolipidoses, rickets), teratogens (retinoic acid, valproic acid, warfarin), hematologic disorders (sickle cell anemia, polycythemia vera), and malformations (microcephaly, holoprosencephaly).2
The most common forms of syndromic craniosynostosis are often the result of de novo autosomal dominant mutations in the fibroblast growth factor receptor (FGFR) and TWIST genes.
Apert syndrome is caused by mutations in the FGFR2 gene on chromosome 10.
Crouzon syndrome is predominantly caused by mutations in FGFR2, although patients with Crouzon syndrome with acanthosis nigricans have a specific mutation in the FGFR3 gene on chromosome 4.
Pfeiffer syndrome is caused by mutations in FGFR1 and FGFR2.
Saethre-Chotzen syndrome is caused by mutations in the TWIST gene located on chromosome 7 but is also associated with mutations in FGFR2.
NATURAL HISTORY
Premature suture fusion restricts growth perpendicular to the suture, thus leading to compensatory growth parallel to the suture that results in characteristic skull deformations.
Sagittal synostosis leads to scaphocephaly, which is characterized by increased anteroposterior (AP) and decreased biparietal width.
Unicoronal and lambdoid synostosis lead to plagiocephaly.
Anterior plagiocephaly is caused by unicoronal synostosis and is characterized by ipsilateral supraorbital rim flattening, forehead recession, deviation of the nasal root to the affected side, temporal retrusion, and vertical orbital dystopia (harlequin deformity).
Posterior plagiocephaly is caused by lambdoid synostosis and is characterized by contralateral bulging and ipsilateral forehead recession.
Metopic synostosis leads to trigonocephaly, which is characterized by a narrow, triangle-shaped forehead, superolateral orbital depression, and hypotelorism.
Bicoronal synostosis leads to brachycephaly, which is characterized by decreased AP diameter and increased biparietal width and height.
Multisuture synostosis typically involving the coronal, lambdoid, and metopic sutures can cause kleeblattschädel or cloverleaf deformity.
Craniosynostosis may also have neurologic, psychiatric, and cognitive implications.3
There is increasing evidence that patients with craniosynostosis have a high incidence of elevated intracranial pressure (ICP).4,5,6
Earlier surgical intervention in craniosynostosis has been shown to be associated with improved neurocognitive outcomes7 and may be mediated by preventing ICP elevation.6
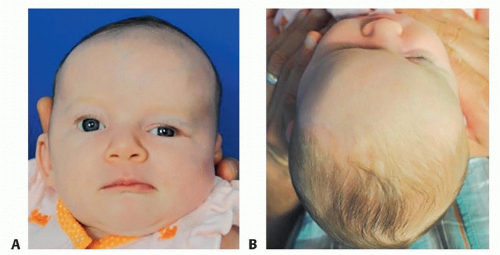
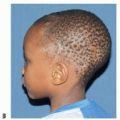
FIG 1 • Female infant with right unicoronal synostosis at 1.5 months of age.
Hydrocephalus is more common in patients with syndromic craniosynostosis. Within the syndromic cohort, Crouzon syndrome patients tend to have the highest incidence of hydrocephalus.
PATIENT HISTORY AND PHYSICAL FINDINGS
Patients will typically present with a history of abnormal head shape during infancy (FIG 1A,B).
Physical exam is the mainstay of diagnosis for single-suture nonsyndromic craniosynostosis.
On exam, patients with single-suture craniosynostosis will have the characteristic skull shapes as described in natural history.
Physical exam should also focus on delineating underlying syndromic etiology, which includes assessing for abnormal facial features and limb anomalies.
Clinical symptoms like headache and visual changes, with papilledema on fundoscopic examination, are signs of increased ICP, a possible sequela of craniosynostosis.
IMAGING
Although physical exam is often sufficient for diagnosis, imaging with either plain radiographs or 3D computed tomography may be required in borderline or complex cases in order to accurately characterize the anatomy and aid in preoperative planning.
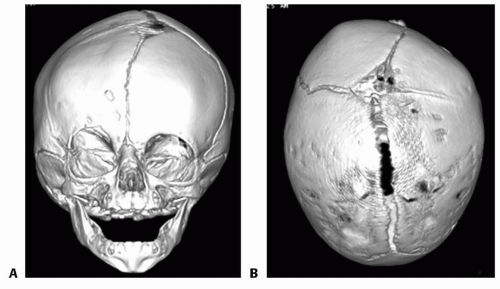
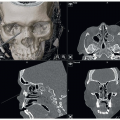
FIG 2 • 3D CT scans of the same female infant with right unicoronal synostosis.
Plain radiographs may be useful if the pretest probability of craniosynostosis (based on history and physical exam) is low.
3D computed tomography is useful for accurately identifying the suture(s) involved and characterizing the bony deformity but can also be used to evaluate the brain parenchyma and other soft tissues (FIG 2).
In infants with suspected syndromic craniosynostosis, genetic testing for the most common genes (FGFR and TWIST) can be performed.
DIFFERENTIAL DIAGNOSIS
Positional plagiocephaly
Congenital torticollis
Craniosynostosis
SURGICAL MANAGEMENT
Craniosynostosis is managed surgically through a variety of different techniques.
The goals of surgery are twofold:
Functional: to prevent intracranial hypertension from damaging the developing brainRelated posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree