Abstract
Erythema multiforme (EM), Stevens–Johnson syndrome (SJS), and toxic epidermal necrolysis (TEN) are immunologically mediated, mucocutaneous reactions triggered by medications and infections. Clinical presentation ranges from targetoid lesions in EM and extensive skin sloughing in TEN. Disease can be self-limited as in EM or potentially fatal as in TEN. Although there is a better understanding of the molecular events leading to the disease state, there remains no standard of care for treatment.
Keywords
Drug rash, Erythema multiforme, EM, Mucocutaneous reaction, Severe drug eruption, Stevens–Johnson syndrome, SJS, Target lesion, Toxic epidermal necrolysis, TEN
- •
Herpes simplex virus infection is a frequent cause of erythema multiforme, while drugs most often cause Stevens–Johnson syndrome and toxic epidermal necrolysis.
- •
Erythema multiforme is often self-limited, skin lesions often target-appearing, and mucosal lesions can occur in the absence of skin lesions.
- •
Stevens–Johnson syndrome patients have systemic illness with prominent mucosal involvement and may have skin sloughing.
- •
Toxic epidermal necrolysis patients have systemic illness and present with painful red skin that progresses to extensive skin sloughing.
- •
Survival for patients with Stevens–Johnson syndrome and toxic epidermal necrolysis improves with early intervention, and the benefit of immunomodulatory therapies is inconclusive.
Erythema multiforme (EM), Stevens–Johnson syndrome (SJS), and toxic epidermal necrolysis (TEN) are immunologically mediated, mucocutaneous reactions triggered by medications and infections. Disease can be self-limited as in EM or potentially fatal as in TEN. The current clinical classification of EM, SJS, and TEN is based on pattern and distribution of skin lesions and maximum extent of skin detachment. However, the clinical classification of these diseases is often debated, and although there is a better understanding of the molecular events leading to the disease state, there remains no standard of care for treatment.
EM is the least severe of the mucocutaneous reactions. It is self-limited, characterized by classic true target lesions—with three zones of inflammation—in the absence of widespread skin sloughing; there is variable mucous membrane involvement, and causality frequently from herpes simplex virus (HSV) or less often other causes. When systemic symptoms or extensive skin sloughing is present, with or without targetoid lesions or mucosal involvement, the diagnoses of SJS and TEN are considered.
Pathogenesis
Immune-mediated pathogenesis has long been suspected for EM, SJS, and TEN. Current concepts center on host immune response to various specific antigenic stimuli from drugs and infections. In EM patients, cutaneous lesions often result from cytotoxic reactions against HSV antigens expressed in epidermal keratinocytes. For the vast majority of patients with SJS and TEN, a cytotoxic reaction results from immunoreactivity against keratinocytes expressing drug-related antigens. Evidence supports a theory that patients are immunogenetically predisposed to developing their disease.
Multiple etiologic agents have been implicated as causes of EM, SJS, and TEN ( Table 11-1 ). The best-documented causes of EM include HSV and Mycoplasma pneumoniae . Fragments of HSV DNA can be found in the cutaneous lesions of EM in more than 80% of cases. While many drugs have been reported to cause EM, such cases may lack classic target lesions and are currently often labeled as mild SJS. Other associations include mononucleosis, other viral infections (e.g., mumps, poliomyelitis, milker’s nodule, and vaccinia), granuloma inguinale, psittacosis, histoplasmosis, syphilis, streptococcal infection, radiation therapy of tumors, sarcoidosis, pregnancy, carcinomas, reticuloses, leukemias, systemic lupus erythematosus (Rowell’s syndrome), and other collagen–vascular diseases. A significant percentage of cases remain idiopathic.
| Infectious agents Herpes simplex Mycoplasma pneumoniae Epstein–Barr virus Mumps Polio Calymmatobacterium Streptococcus Vaccinia Yersinia Tuberculosis Treponema pallidum Chlamydia Deep mycoses (e.g., histoplasmosis) Dengue virus Cytomegalovirus |
| Medications Sulfonamides Sulfasalazine NSAIDs Carbamazepine Phenytoin Lamotrigine Barbiturates Allopurinol Penicillins Cephalosporins Quinolones Tetracyclines Contrast agents Nevirapine |
| Other conditions Irradiation of tumors Immunizations Connective tissue disease (e.g., systemic lupus erythematosus) Sarcoidosis Inflammatory bowel disease Pregnancy |
Although there is a complex interplay between host immune cells, CD8 + cytotoxic T lymphocytes and natural killer cells appear mainly responsible for the keratinocyte apoptosis seen in the mucocutaneous reactions. The mechanism of immune cell activation is theorized to be secondary to the pro-hapten concept, p-i concept, or both. In the pro-hapten concept drug metabolites bind to cellular peptides creating a highly immunogenic molecule capable of immune system activation. In the p-i concept the drug or drug metabolite can bind directly to MHC or T-cell receptors and stimulate an immune response.
Several MHC I allotypes have been associated with increased incidence of SJS and TEN. Han Chinese, Thai, Malaysian, and South Indian populations with MHC I allotype (HLA)-B ∗ 1502 have increased risk of SJS/TEN from aromatic antiepileptic agents such as phenytoin, carbamazepine, lamotrigine, and oxcarbazapine. Likewise, populations of European descent with allotype HLA-B ∗ 5801 have increased incidence of allopurinol-induced SJS/TEN. These allotypes may be genetic markers or could be involved in the pathogenesis of the mucocutaneous syndromes. The US Food and Drug Administration recommends that patients of East Asian descent have testing for the HLA-B ∗ 1502 genotype prior to receiving carbamazepine.
The crucial mediator of keratinocyte apoptosis in the mucocutaneous reactions appears to be granulysin, a molecule found in the cytotoxic granules of CD8 + , NK, and NK/T cells, which is capable of causing membrane instability and destruction resulting in cell death. Increased levels of granulysin are found in TEN blisters as well as serum and correlate with disease severity. Multiple other factors are implicated in the disease pathogenesis of SJS and TEN as well, including death receptor Fas (CD95), soluble Fas ligand (FasL) produced by peripheral blood mononuclear cells, and FasL on epidermal keratinocytes, cytotoxic T cells, and NK cells. Interaction of Fas and FasL induces signaling, which rapidly leads to apoptosis. Antibodies present in pooled human intravenous immunoglobulins (IVIg) may block Fas-mediated keratinocyte death, and use of IVIg for treatment of TEN rests on this theory. Markers of oxidative stress are also elevated in TEN patients, and may result from TNF-α- and IFN-γ-induced production of nitric oxide within inflammatory cells and keratinocytes. Other inflammatory mediators found elevated in TEN patients include alarmins, TRAIL (TNF-related apoptosis-inducing ligand), TWEAK (TNF-related weak apoptosis inducer), alpha-defensins, and granzyme B and perforin released by cytolytic T cells.
Clinical Manifestations
The current classification of EM, SJS, and TEN is based on consensus definitions published in 1993. An international group reviewed hundreds of historical cases and agreed upon clinical definitions based on pattern and distribution of skin lesions and maximum extent of skin detachment. The consensus group determined that EM differed significantly enough from SJS and TEN that it represented its own clinical group, and SJS and TEN were variants of a single disease. However, while there is general agreement that EM represents a different disease, many believe that within the SJS and TEN spectrum exist two distinct diseases with different clinical presentations. Additionally, a mycoplasma-induced rash and mucositis syndrome may exist, distinct from EM/SJS and characterized by a mild disease course. Thus, the diagnosis of EM, SJS, and TEN is often debated and different clinical classifications exist.
Erythema Multiforme
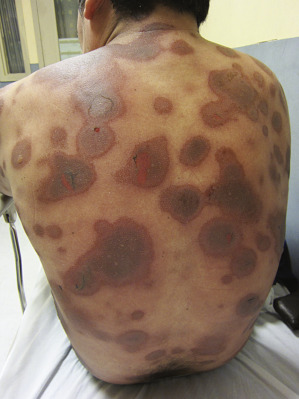
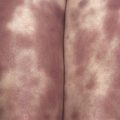
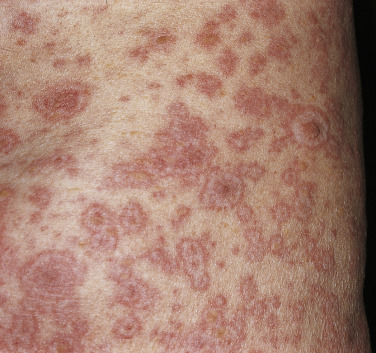
EM begins with either a mild prodrome of malaise and low-grade fever or no systemic involvement at all. Skin lesions are often target-appearing ( Fig. 11-1 ) with three concentric zones, most commonly: (1) a central dusky area, (2) a middle pink or edematous zone, and (3) an outer red ring. Although target lesions are characteristic of EM, lesions need only be monomorphic for an EM diagnosis, and target lesions themselves are not diagnostic for EM. Target lesions can be seen in viral-induced eruptions and may not show the characteristic histology necessary to confirm a diagnosis of EM. Lesions may be asymptomatic to burning or pruritic, last from 1 to 2 weeks, and tend to evolve to have a dusky appearance. Mucosal surfaces may be involved in EM ( Fig. 11-2 ), and mucosal involvement can occur in the absence of skin lesions. Within EM, two variants have been observed and studied: HSV-associated cases with target lesions predominantly on the extremities and drug-associated cases with target lesions in a diffuse or central pattern. The distinct clinical presentations of EM appear to have different molecular mechanisms, as HSV-associated EM lesions express IFN-γ and drug-induced EM lesions express TNF-α. Recurrent EM is not uncommon, and is often due to HSV reactivation.
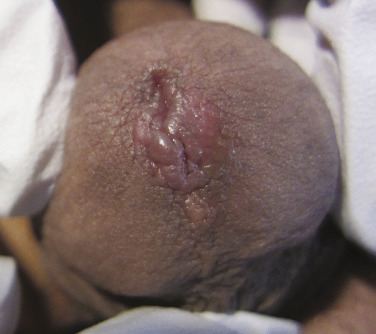
Stevens–Johnson Syndrome
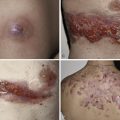
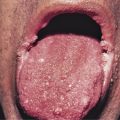
The consensus group in 1993 suggested that SJS is a less severe variant of TEN, only differing by percent of skin involvement and not on clinical presentation. SJS was defined to involve less than 10% denudation of the total body surface area. Although this is a widely accepted definition, other opinion suggests SJS diagnostic criteria based on clinical disease presentation rather than resulting percent of skin sloughing. The classification we favor suggests that SJS presents with (1) a prodrome of fever and malaise for 1 day to 2 weeks, (2) two or more sites of mucous membrane involvement, and (3) skin involvement that may include characteristic true target lesions, targetoid lesions (with two zones of inflammation, such as central nonblanching redness surrounded by blanching erythema), or other monomorphic lesions that may progress to skin denudation that is indistinguishable from TEN ( Fig. 11-3 ). This definition emphasizes presenting symptoms and mucosal involvement and is similar to the original case descriptions of Stevens and Johnson in 1922 and Thomas in 1950 where patients had prodromal symptoms, mucosal involvement that was often ocular and oral, and a generalized skin eruption. Mucosal lesions of SJS may affect the ocular conjunctivae ( Fig. 11-4 ), oral cavity ( Fig. 11-5 ), vaginal mucosa or penis meatus, anal mucosa, and esophagus. Mucosal complications may include keratitis, conjunctival scarring, uveitis, scleral perforation, urethral stricture, vaginal scarring, and esophageal stricture. Patients may experience fevers, arthralgias, arthritis, myalgias, hepatitis, bronchopulmonary disease, glomerulonephritis, or acute renal tubular necrosis.