Abstract
Many diseases with cutaneous manifestations are also associated with monoclonal gammopathies. Monoclonal gammopathies are often idiopathic and increase in incidence with age and by ethnicity: African-American > Caucasian > Hispanic. This chapter addresses those diseases that are directly related to the monoclonal protein (e.g., cryoglobulinemia, cold agglutinins, Waldenström macroglobulinemia), are associated with the presence of a monoclonal protein at a much higher than chance association (e.g., POEMS syndrome, scleromyxedema, necrobiotic xanthogranuloma), as well as the amyloidoses, some of which are directly related to monoclonal protein, and which must be differentiated from other causes of amyloid protein deposition in the skin.
Keywords
Amyloidoses, Cold agglutinins, Cryofibrinogenemia, Cryoglobulinemia, Cutaneous plasmacytoma, Gammopathy, Necrobiotic xanthogranuloma, POEMS syndrome, Scleromyxedema, Waldenström macroglobulinemia, Waldenströn hypergammaglobulinemic purpura
- •
The presence of a monoclonal gammopathy may be an incidental finding, but there are many cutaneous disorders that are due to or are strongly associated with the presence of a monoclonal gammopathy.
- •
A usual way to organize thinking around the syndromes divides them into diseases directly related to the monoclonal protein, diseases frequently associated with a monoclonal protein without a clear causative role, and disorders of amyloid deposition, the most common systemic form being due to light-chain synthesis into amyloid protein.
- •
The diagnosis and treatment of these disorders will be addressed within this framework.
A wide variety of diseases associated with monoclonal immunoglobulin or light-chain production may cause cutaneous lesions, and these will be addressed by category: those directly related to monoclonal protein (e.g., cryoglobulin), those frequently associated with gammopathy (e.g., scleromyexedema), and those resulting from abnormal metabolism of monoclonal proteins (e.g., light-chain-related amyloidosis).
Commonly, monoclonal immunoglobulin production is simply a monoclonal gammopathy of undetermined significance (MGUS). MGUS is defined as a serum monoclonal protein <30 g/L; <10% plasma cells in the bone marrow; and the absence of end-organ damage (hyper c alcemia, r enal insufficiency, a nemia, and/or b one lesions [CRAB]). Although this disorder is uncommon in young individuals, its incidence increases with age, reaching 3% in those 50 years of age or older, and 5% in those over 70 years. The risk of progression to multiple myeloma or a related disorder is 1% per year. Three risk factors are useful in predicting the likelihood of progression (as measured at 20 years’ follow-up): (1) an increase in serum-free light chains; (2) an MGUS of nonimmunoglobulin G (IgG) origin; or (3) a serum M protein of 15 g/L or more. The risk of multiple myeloma at 20 years was 58% with all three risk factors, 37% with two risk factors, 21% with one risk factor, and 5% when no risk factors were present. Smoldering myeloma is a term used to describe an asymptomatic phase of myeloma characterized by a serum IgG or IgA monoclonal protein >30 g/L and/or >10% plasma cells in the bone marrow, but no evidence of myeloma-related end-organ damage. In this group, the cumulative probability of progression to active multiple myeloma or amyloidosis was 51% at 5 years, 66% at 10 years, and 73% at 15 years, with a median time to progression of 4.8 years.
Disorders Directly Related to Monoclonal Proteins
Pathogenesis
Monoclonal proteins may cause disease directly by acting as cryoglobulins, by raising serum viscosity, or by acting as cold agglutinins. Cryoglobulins are immunoglobulins with temperature-dependent conformational change leading to water insolubility and precipitation on exposure to cold. They may be unstable in other settings, such as in the hyperosmotic environment found in the kidneys, or in microvascular areas with a slow blood flow. The most critical factor that determines the behavior of the cryoglobulin in vivo is the temperature at which it begins to precipitate. If that temperature approaches those found in the cutaneous microvasculature on cold exposure, cold-induced disease is usually significant. If it precipitates only at a temperature well below room temperature, symptoms, if any, will not be cold-related. Cryoglobulins are divided into three categories, depending on their composition. Type I cryoglobulins consist of a single monoclonal protein; type II are composed of a monoclonal immunoglobulin with anti-IgG (rheumatoid factor) activity that binds to polyclonal serum IgG; and type III cryoglobulins consist of polyclonal immunoglobulins, usually with anti-IgG activity, that bind to polyclonal serum IgG (a polyclonal rheumatoid factor). Types I and II cryoglobulinemia are often (but not always) associated with a lymphoproliferative disorder or plasma cell dyscrasia. Patients with type II cryoglobulinemia may have IgM, IgG, or IgA as their monoclonal rheumatoid factor. Only IgM rheumatoid factor testing is routinely available. Since the monoclonal protein is bound to a polyclonal IgG antigen, serum protein electrophoresis may not show a discrete M-spike, and if the specimen is allowed to cool before sampling, all studies will be negative.
The hyperviscosity syndrome results from a significant increase in whole blood viscosity. Such an increase may be related to an increase in the cellular elements in the blood, as in polycythemia vera, but most often results from a change in serum viscosity due to large amounts of monoclonal protein in the blood.
Cold agglutinin disease is a cold antibody-induced autoimmune hemolytic anemia. The antibody, usually IgM, binds to the red cell in the cold and initiates complement activation; it then elutes at body temperature while the complement activation proceeds to red cell lysis. The cold agglutinin also promotes temperature-dependent agglutination of red cells, leading to sludging or occlusion of the blood flow in the microvasculature exposed to cold temperatures.
Clinical Manifestations
Cryoglobulinemia
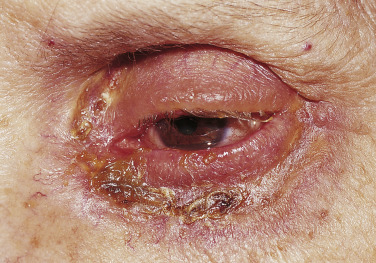
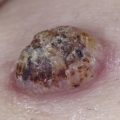
Roughly 5% to 10% of myeloma proteins and macroglobulins are cryoprecipitable. Type I cryoglobulins are usually IgM and therefore primarily intravascular; however, they can also be composed of IgG. To be symptomatic they should precipitate at temperatures easily attained in the cutaneous microvasculature. Such disease may present as Raynaud’s phenomenon, livedo reticularis, digital infarcts, peripheral gangrene, or purpura; the latter may be palpable ( Fig. 21-1 A and B ), exhibit central necrosis, or have a livedoid or retiform component ( Fig. 21-1 C ).
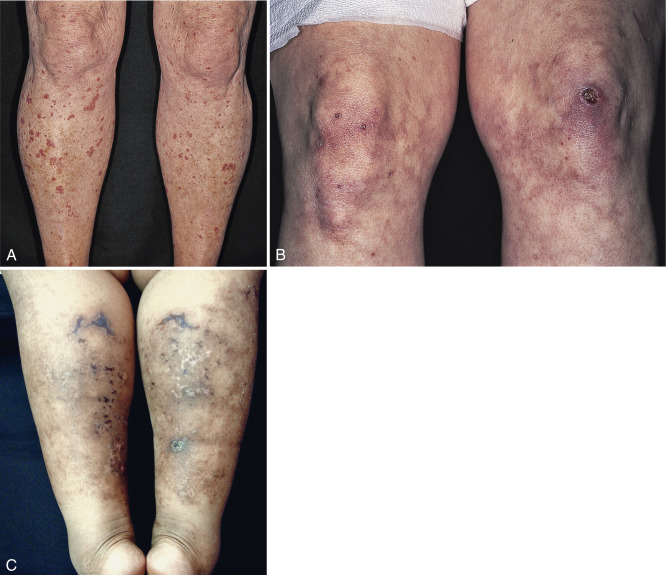
Cold sensitivity is more variable with types II and III cryoglobulins because these are usually bound to normal IgG. The classification of essential mixed cryoglobulinemia is now known to include primarily patients with hepatitis C viral infection. Mixed cryoglobulins may be detected as cryoproteins or as rheumatoid factor in the laboratory; in the patient, they are more likely to cause disease as an immune complex than as a cryogelling protein. For this reason, mixed cryoglobulinemias tend to present with features of leukocytoclastic (necrotizing) vasculitis that affect both small- and medium-sized vessels in the skin and elsewhere. Palpable purpura, digital infarcts, arthralgias and arthritis, and glomerulonephritis are the usual clinical features. Some patients with an underlying lymphoproliferative disease may develop angioedema with urticaria as a result of C1 esterase inhibitor depletion; the monoclonal protein in such patients may also behave as a cryoglobulin in the test tube. Finally, some cases of cold-induced urticaria are caused by a circulating cryoglobulin without evidence of any associated disease.
Hyperviscosity Syndrome
Hyperviscosity syndrome may present as macular hemorrhage, mucous membrane bleeding, retinopathy, neurologic disturbances, hypervolemia, or cardiac failure, and, if caused by a cryoglobulin, Raynaud’s phenomenon. Symptoms require a four- to fivefold increase in blood viscosity, usually associated with >3 g/dL IgM (or >15 g/dL IgG, >4 to 5 g/dL polymerized IgG3, >10 to 11 g/dL IgA, >6 to 7 g/dL polymerized IgA) and can also be seen with chylomicronemia syndrome, and with cellular causes of hyperviscosity in polycythemia vera, sickle cell disease, leukemia, and spherocytosis. Hyperviscosity alone may not fully explain the increased bleeding tendency in these patients. Additional factors in some patients are antibody activity against clotting factors, platelet dysfunction as a result of surface coating by immunoglobulin, and other poorly understood clotting defects.
Cold Agglutinin Disease
Cold agglutinin disease is characterized clinically by episodes of hemolytic anemia, hemoglobinuria, and cold-mediated vaso-occlusive phenomena. Patients may develop acrocyanosis, Raynaud’s-like phenomenon, or generalized livedo reticularis, but cutaneous ulcerations or necrosis are unusual. Jaundice or pallor may follow a severe episode of hemolysis. Cold agglutinin disease occurs in two forms: primary (idiopathic) and secondary. Patients with primary cold agglutinin disease may develop features diagnostic of Waldenström’s macroglobulinemia, whereas secondary forms follow certain infections. Cold agglutinins can also be categorized by their red cell antigen affinity. The antibodies are usually directed against the I/i antigen system, and, rarely, against Pr group antigens. Anti-I antibodies occur primarily in association with idiopathic disease, Mycoplasma pneumonia, and some lymphomas; anti-i-specific antibodies are associated with infectious mononucleosis and some lymphomas. The cold agglutinins are monoclonal in primary and lymphoma-associated disease and polyclonal in postinfectious disease. The rare cases of IgA cold agglutinin disease are also characterized by red cell agglutination in the microvasculature, but hemolytic anemia does not develop because the cell-bound IgA does not fix complement.
Waldenström’s Macroglobulinemia
This is a disease characterized by a serum monoclonal IgM spike and by malignant lymphoplasmacytoid proliferation, primarily in the bone marrow, liver, spleen, and lymph nodes. In some patients the IgM paraprotein may behave as a monoclonal (type I) cryoglobulin or a cold agglutinin. Urticarial vasculitis associated with an IgM monoclonal protein, bone pain with hyperostosis, and intermittent fever is known as Schnitzler syndrome.
Cryofibrinogenemia
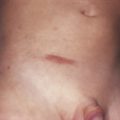
Cryofibrinogen is a plasma complex of fibrin, fibrinogen, and fibronectin that can precipitate on cooling and clot when combined with thrombin. Cryofibrinogenemia occurs as a primary disorder, or as an associated disorder in patients with neoplasia, acute infections, collagen vascular disorders, or thromboembolic disease. Patients with cryofibrinogenemia may present with recurrent painful cutaneous ulcerations of the lower leg and foot. Purpura, often nonpalpable, may accompany the ulcers, which are usually small. The ulcerations heal with ivory stellate scars, resembling the cutaneous features of livedoid vasculopathy ( Fig. 21-2 ).
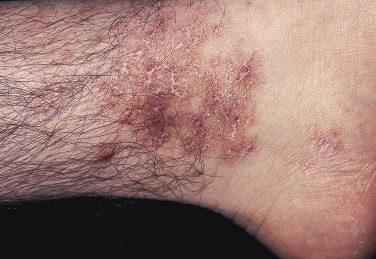
Differential Diagnosis
In cases of mixed cryoglobulinemia presenting as leukocytoclastic vasculitis, the differential diagnosis primarily includes other necrotizing vasculitides. Hyperviscosity syndrome is usually caused by a clonal lymphoproliferative disorder, but it can result from other hematologic disorders, such as polycythemia vera. Raynaud’s phenomenon may be idiopathic (Raynaud’s disease), or may be secondary to an autoimmune connective tissue disease, including scleroderma, lupus erythematosus, rheumatoid arthritis, or Sjögren’s syndrome. Other diseases may resemble Raynaud’s phenomenon; in addition to those mentioned in this section, conditions such as ergotism, Buerger’s disease, chilblains (pernio), and acrocyanosis should be considered. In patients presenting with lesions of dermal vessel occlusion, disorders that must also be considered are many (see Chapter 15 ).
Histopathologic Findings
Early cutaneous lesions of monoclonal cryoglobulinemia demonstrate intravascular amorphous eosinophilic material composed principally of precipitated cryoglobulin. There is often red blood cell extravasation into the dermis. Inflammation is usually minimal, and appears to be a response to, rather than a cause of, necrosis, which results from vessel occlusion. Biopsy of a late lesion of occlusion with necrosis may show changes of secondary leukocytoclastic vasculitis. By contrast, early cutaneous lesions of mixed cryoglobulinemia show histopathologic changes of leukocytoclastic (necrotizing) vasculitis. Immunoreactant deposition in vessel walls can be seen on direct immunofluorescence microscopy.
Cutaneous lesions that are the result of cryofibrinogenemia, cold agglutinin disease, or paroxysmal nocturnal hemoglobinuria should show histologic evidence of multiple dermal vessel thrombosis, usually in the absence of significant inflammatory infiltrate.
Evaluation
Although the detection and analysis of a serum or urine monoclonal immunoglobulin or Bence Jones protein (i.e., light chain) was classically accomplished via serum or urine protein electrophoresis, immunofixation techniques have proved to be significantly more sensitive. The serum-free light-chain assay has emerged as the most sensitive method for the detection of plasma cell dyscrasias characterized by overproduction of light chains. When a patient is suspected of having a cryoprotein-related disease, blood can be drawn for both serum and plasma sampling and kept at body temperature until the cellular elements have been removed. Following at least overnight refrigeration, each sample is then examined for evidence of a cryoprecipitate. A cryoglobulin should appear in both the serum and the plasma sample, whereas cryofibrinogen will appear in only the plasma fraction. Immunofixation of the precipitated cryoglobulin can then define its composition. Patients found to have type I or II cryoglobulins should be examined for an underlying plasma cell dyscrasia or lymphoproliferative disorder. Patients with type II or III cryoglobulins should be evaluated for hepatitis C viral infection; nonhepatitis-related disease may be secondary to an underlying connective tissue or autoimmune disease, or to other chronic infections or inflammatory diseases.
Vessel occlusion caused by cold agglutinins is rare, despite their low-level presence in many different acute and chronic diseases. If hyperviscosity syndrome is suspected, serum viscosity measurements are usually easily obtained from clinical laboratories, but whole-blood viscosity measurements remain a specialized research procedure.
Treatment
The treatment of cryoglobulinemia depends on the clinical features. If clinically relevant cold sensitivity is present, adequate clothing and avoidance of cold exposure are essential and may be all that is required. For those with more severe disease, treatment of the underlying plasma cell dyscrasia or lymphoproliferative disorder is indicated. In patients with type II or III cryoglobulinemia, in whom necrotizing vasculitis is the usual presenting finding, cutaneous lesions may respond to oral dapsone or colchicine therapy. Aggressive therapy with systemic corticosteroids in combination with immunosuppressive or cytotoxic agents may be required in severe systemic disease. In hepatitis-C-related cryoglobulinemia, interferon regimens of antiviral therapy may reduce the vasculitis but can occasionally induce flaring. Published data are rare with regard to the effect of new specific antihepatitis C agents; vasculitis remission is reported. Patients with specific underlying etiologic disorders may respond to effective treatment of that disorder. Plasmapheresis may provide temporary but rapid relief of symptoms in patients with high levels of circulating cryoglobulins, and it may be synergistic when combined with chemotherapy in appropriate cases. Intravenous γ-globulin therapy is occasionally successful in cryoglobulinemia and refractory vasculitis.
Cold agglutinin disease is best treated by keeping the patient (particularly the extremities) warm. Cytotoxic agents may be useful in some patients, but plasmapheresis is contraindicated because it may induce severe hemolytic anemia. Postinfectious cases of cold agglutinin disease are usually self-limiting.
For patients in whom simple measures do not address the clinical manifestations of cryofibrinogenemia, the use of an oral anabolic steroid such as stanozolol (4 to 8 mg/day) or danazol may prove beneficial, with rapid pain relief and ulcer healing observed in a number of patients. Stanozolol and danazol are androgenic steroids with fibrinolytic properties. Stanozolol has been removed from the market in the United States, but apparently can be obtained through compounding pharmacies. Other therapies for livedoid vasculopathy associated with cryofibrinogenemia include heparin, warfarin, streptokinase, plasmapheresis, immunosuppressive agents, and tissue plasminogen activator.
Disorders Associated with Monoclonal Protein Production
Pathogenesis
This group of disorders is united by the frequent finding of an associated monoclonal gammopathy. Some diseases are almost always associated with a monoclonal gammopathy (e.g., POEMS syndrome, scleromyxedema), whereas others are frequently associated with a monoclonal gammopathy (e.g., normolipemic plane xanthoma). Many diseases have a greater than chance association with the presence of a serum or urine monoclonal protein, but this is not required for typical disease expression (e.g., scleredema).
Clinical Manifestations
POEMS Syndrome
This was recognized as a distinct entity in Japan in 1968 and is also known as the Crow–Fukase or Takatsuki syndrome. POEMS is an acronym for polyneuropathy, organomegaly, endocrinopathy, M-protein, and skin changes. In a large series men were affected twice as frequently as women, and patients were young to middle-aged adults with a mean age of 46 years (compared to mean age of myeloma presentation of 62 years). All patients had peripheral polyneuropathy (usually sensorimotor), 97% had elevated cerebrospinal fluid protein, and 62% had papilledema. Organomegaly was manifested as hepatomegaly in 82% of the patients, lymphadenopathy in 65%, and splenomegaly in 39%. The most common endocrine abnormalities were impotence (78%) and gynecomastia (68%) in men, and amenorrhea (68%) in women. Additional endocrine findings in this and other series include glucose intolerance (28% to 48%), hyperthyroidism (10% to 24%), and hyperprolactinemia, adrenal insufficiency, or hypercalcemia (rare).
Most patients (75%) have had a serum or rarely a urine monoclonal spike. Of these spikes, approximately 55% were IgG1 and 40% were IgA1. Although in some of these patients the disease may ultimately progress to multiple myeloma, this does not always occur. Slightly more than half of patients with POEMS syndrome had bone lesions, and 85% of patients with bone lesions had osteosclerotic lesions, with or without osteolytic lesions; this is in contrast to a large series of patients with myeloma in whom osteosclerotic lesions comprised only 0.5% to 3.0% of bone lesions. Also, unlike the findings in osteolytic multiple myeloma, anemia, hypercalcemia, and renal insufficiency are uncommon, and extensive bone marrow infiltration by plasma cells is rare.
Cutaneous changes are common in this disorder, and reported changes include diffuse hyperpigmentation (93% to 98%); peripheral edema (92%); and sometimes anasarca, hypertrichosis (78% to 81%), a poorly characterized skin thickening (77% to 85%), and digital clubbing (56%). Cutaneous angiomas occur in 24% to 44% of patients and include cherry, verrucous, subcutaneous, or “glomeruloid” angiomas ( Fig. 21-3 ). The histopathological finding of a glomeruloid angioma is highly indicative of this syndrome. That circulating levels of vascular endothelial growth factor (VEGF) are markedly increased in these patients may provide some explanation for the vascular changes in POEMS. This increase in VEGF may come from aggregating platelets overly rich in VEGF, which could provide very high local microcirculatory concentrations of VEGF. In patients with POEMS in association with multicentric Castleman’s disease, viral interleukin (IL)-6 produced by human herpesvirus-8 could also lead to increased circulating levels of VEGF. Sclerodermoid changes, facial atrophy, flushing, Terry’s nails, acrocyanosis, Raynaud’s phenomenon, and sicca syndrome have also been described. Biopsy findings of the skin are often nonspecific, but include hyperpigmentation, dermal thickening caused by edema and an increase in collagen and proteoglycan, microvascular proliferation, and occasional large fibroblasts in the dermis.
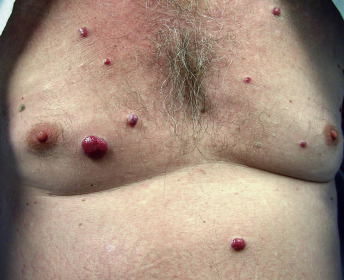
Variable findings include ascites, pleural effusions, fever, polycythemia, leukocytosis, thrombocytosis, and an elevated erythrocyte sedimentation rate. POEMS syndrome may predispose to arterial and venous thromboses and stroke. Associations with Castleman’s disease have been reported, as have presentations such as flushing, hypotension, and bronchial spasm, mimicking carcinoid. The AESOP (adenopathy and extensive skin patch overlying a plasmacytoma) syndrome describes a distinctive presentation of a slowly extending violaceous skin patch overlying a solitary plasmacytoma of bone, associated with enlarged regional lymph nodes. Of the four reported patients, all had neuropathy and two developed POEMS syndrome.
Primary Cutaneous Marginal Zone Lymphoma of Mucosa-Associated Lymphoid Tissue Type (PCMZL-MALT): Formerly Cutaneous Plasmacytoma
In the WHO-EORTC and current WHO classifications, cutaneous plasmacytomas and cutaneous immunocytomas are considered to be variants of PCMZL. These are rare and may be solitary or multiple. Most of these cutaneous lesions are smooth, nontender, cutaneous, or subcutaneous nodules, skin-colored to violaceous, and 1 to 5 cm in diameter, and they may be crusted or ulcerated. They are usually located on the trunk, extremities, or face. All immunoglobulin classes have been associated with PCMZL, but most are IgG- or IgA-producing cells. True cutaneous plasmacytomas indicate a large tumor cell burden in patients with multiple myeloma, and therefore usually occur late in the course of the disease, either as an extension from underlying bone or as distinct cutaneous metastases. Although IgD myeloma is rare, patients with this disease have a higher incidence of extramedullary lesions, including cutaneous plasmacytomas (up to 18%). The IgD subset of myeloma usually develops in young men and has an aggressive course.
A lesion of PCMZL may be an isolated finding, even with long-term follow-up. Because the number of plasma cells in such a lesion is small and the amount of immunoglobulin synthesized is directly related to cell numbers, such patients are unlikely to have a serum monoclonal antibody spike. Conversely, the presence of a monoclonal spike suggests extracutaneous disease.
Cutaneous and Systemic Plasmacytosis
Plasma cell-rich lesions have been described in cutaneous and systemic plasmacytosis as well as plasma cell orificial mucositis. Cutaneous and systemic plasmacytosis is a rare disorder reported almost exclusively in patients of Japanese descent, and is characterized by widespread reddish-brown macules (due to polyclonal plasma cell infiltration), polyclonal hypergammaglobulinemia, peripheral adenopathy (∼60%), and sometimes infiltration of the lung, liver, spleen, or kidneys. Although rare, these disorders present with so extensive an infiltration of plasma cells that, on biopsy, they may mimic cutaneous plasmacytomas.
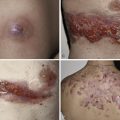
Necrobiotic Xanthogranuloma with Paraproteinemia
This is a distinctive entity, typically presenting with yellowish periorbital plaques or nodules that tend to ulcerate and heal with scar formation. Lesions may also develop on the trunk or proximal limbs, especially in flexural areas ( Fig. 21-4 ). The lesions often extend deeply into the dermis and subcutis and, on biopsy, show both Touton and foreign body giant cells, broad areas of altered collagen, and necrobiosis on a background of extensive granulomatous inflammation. Most patients have a monoclonal gammopathy (IgG, often κ), many have leukopenia, bone marrow plasmacytosis is common, but multiple myeloma or other lymphoproliferative disorders are rare. In one series, hepatomegaly or splenomegaly was reported in 20 of 48 patients.