The visible pigmentation of the skin or hair is a combination of the amount of melanin, type of melanin (eumelanin vs. pheomelanin), degree of vascularity, presence of carotene, and thickness of the stratum corneum. Other materials can be deposited abnormally in the skin, leading to exogenous pigmentation. Eumelanin is the primary pigment producing brown coloration of the skin. Pheomelanin is yellow or red and is also produced solely in melanocytes. Melanin is formed from tyrosine, through the action of tyrosinase, in the melanosomes of melanocytes. A multitude of genes are expressed only in melanosomes and are important in melanin production and delivery. Melanosomes are lysosome-related organelles (LROs). Melanosome formation and the end result, pigmentation, require both the adequate manufacture of melanin and the appropriate transport of melanosomes within the melanocyte. The melanosomes are transferred from a melanocyte to a group of 36 keratinocytes called the epidermal melanin unit, to which they provide melanin. The variations in skin color between people are related to the degree of melanization of melanosomes, their number, and their distribution in the epidermal melanin unit. Disorders of loss or reduction of pigmentation may be related to loss of melanocytes or the inability of melanocytes to produce melanin or transport melanosomes correctly. Wood’s light examination is often performed to evaluate lesions of hyperpigmentation or hypopigmentation. Hyperpigmented lesions that enhance with Wood’s light usually have increased epidermal melanocyte number or activity. If the lesions do not enhance, the melanin is located in the dermis. Wood’s light will greatly enhance depigmented lesions (complete loss of pigment) but does not enhance lesions with partial pigment loss (hypopigmentation).
Pigmentary Demarcation Lines
Pigmentary demarcation boundaries of the skin can be classified into groups based on their anatomic location, orientation, and degree of pigmentation (hyperpigmentation or hypopigmentation):
- 1.
Group A: lines along the outer upper arms with variable extension across the chest
- 2.
Group B: lines along the posteromedial aspect of the lower limb
- 3.
Group C: paired median or paramedian lines on the chest, with midline abdominal extension
- 4.
Group D: medial, over the spine
- 5.
Group E: bilaterally symmetric, obliquely oriented, hypopigmented macules on the chest
- 6.
Groups F, G, and H: facial pigmentary demarcation lines
The term acquired, idiopathic, patterned facial pigmentation (AIPFP) has been used to encompass pigmentary demarcation lines of the face, as well as idiopathic periorbital and perioral pigmentation. These are patterned, bilateral, and homogeneous and have various shades of brown with a variable gray undertone. Periorbital and perioral hyperpigmentation occur in the late teens and early twenties. Other forms of facial hyperpigmentation occur later (average age >30). Periorbital pigmentation usually is demarcated by a band of normal skin beneath the upper eyebrow superiorly and the orbital rim inferiorly. It may extend outward onto the lateral cheek or over the root of the nose. One third of patients with perioral pigmentation have a family history. These patterns of facial pigmentation may represent variations of embryologic pigmentation.
Pigmentary demarcation lines must be distinguished from the much rarer condition, acquired dermal melanocytosis (ADM). This primarily affects Asian and Hispanic women (male/female ratio 1 : 17). The face is the most common location, and it includes the entities bilateral and unilateral nevus of Ota–like macules (Hori and Sun nevus, respectively). ADM can first appear during pregnancy or therapeutic use of estrogen/progesterone. Lesions present as blue-gray patches, with superimposed brown macules. Infrequently, the trunk or extremities may be affected. Lesions do not enhance with Wood’s light. They may be localized (after trauma) or may be more diffuse. Ultraviolet (UV) light and psoralen plus UVA (PUVA) therapy are possible precipitants. Biopsy shows melanocytes in the dermis, similar to the findings in mongolian spot, nevus of Ota, and nevus of Ito. The lesions appear to represent activation of melanin production by residual dermal melanocytes because biopsies in “normal” skin adjacent to the pigmented lesions show dermal melanocytosis.
Cho E, et al: Type B pigmentary demarcation lines of pregnancy involving the anterior thighs and knees. Ann Dermatol 2012; 24: 348.
Chuah SY, et al: Acquired dermal melanocytosis of the nose. J Eur Acad Dermatol Venereol 2015; 29: 827.
Fauconneau A, et al: Acquired dermal melanocytosis of the back in a Caucasian woman. Am J Dermatopathol 2012; 34: 562.
Mintz S, Velez I: Pigmentary demarcation lines (Futcher lines). Quintessence Int 2010; 41: 873.
Nagase K, et al: Acquired dermal melanocytosis induced by psoralen plus ultraviolet A therapy. Acta Derm Venereol 2012; 92: 691.
Peck JW, Cusack CA: Futcher lines. Cutis 2013; 92: 100.
Permatasari F, et al: Late-onset acquired dermal melanocytosis on the hand of a Chinese woman. Indian J Dermatol Venereol Leprol 2013; 79: 269.
Purnak S, et al: Pigmentary demarcation lines of pregnancy in two Caucasian women. J Eur Acad Dermatol Venereol 2015; 29: 2058.
Sarma N, et al: Acquired, idiopathic, patterned facial pigmentation (AIPFP) including periorbital pigmentation and pigmentary demarcation lines on face follows the lines of Blaschko on face. Indian J Dermatol 2014; 59: 41.
Xu C, et al: Types E and C of pigmentary demarcation lines in two Chinese sisters. Eur J Pediatr Dermatol 2014; 24: 71.
Abnormal Pigmentation
Hemosiderin Hyperpigmentation
Pigmentation resulting from deposits of hemosiderin occurs in purpura, hemochromatosis (see section later in chapter), hemorrhagic diseases, and stasis dermatitis. Clinically, hemosiderin hyperpigmentation is distinguished from postinflammatory dermal melanosis by a golden-brown hue, unlike the brown or gray-blue pigmentation of epidermal or dermal melanin, respectively. At times, a biopsy is required to distinguish melanin-induced from hemosiderin-induced hyperpigmentation. Extravasation of iron into the soft tissue from a poorly functioning venous catheter can cause local hemosiderosis of a limb. Multiple transfusions (>20) can result in cutaneous iron deposits in about 20% of patients. Drinking tea while ingesting an iron-containing solution can result in iron staining of the tongue and teeth, simulating black hairy tongue.
Medication Pigment
Some medications, including minocycline, deposit in the skin and complex with both iron and melanin, making uniquely colored (usually blue-gray) deposits.
Postinflammatory Hyperpigmentation (Postinflammatory Pigmentary Alteration)
Any natural or iatrogenic inflammatory condition can result in hyperpigmentation or hypopigmentation ( Fig. 36.1 ). Postinflammatory dyspigmentation is more common in persons with Fitzpatrick skin types IV, V, and VI, especially types IV and V. It is more likely to occur after laser treatment when performed in premenstrual women. It affects both genders equally.
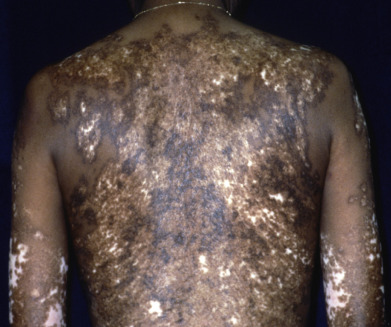
Hyperpigmentation may result from the following two mechanisms:
- 1.
Increased epidermal pigmentation via increased melanocyte activity
- 2.
Dermal melanosis from melanin dropout from the epidermis into the dermis
Wood’s light examination will distinguish these two patterns of postinflammatory hyperpigmentation. Lesions of hyperpigmentation tend to be tan to brown and may have a gray hue, caused by dermal melanin ( Fig. 36.2 ).
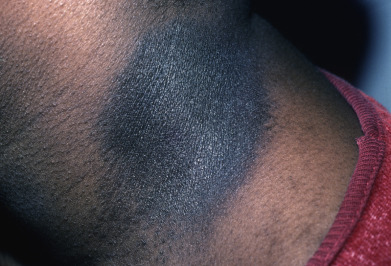
Hypopigmented lesions are prominently lighter than the surrounding area. Histologically, there is melanin in the upper dermis and around upper dermal vessels, located primarily in macrophages (melanophages). The pattern of the dermal melanosis does not predict whether the lesion will be lighter or darker as a result of the prior inflammatory process—thus the tendency of pathologists to provide a diagnosis of “postinflammatory pigmentary alteration” (PIPA) in such cases.
Postinflammatory dyspigmentation will often resolve on its own as long as the process that lead to the dyspigmentation (such as acne) does not continue and replace those areas that have normalized. Postinflammatory hypopigmentation (such as pityriasis alba) should be treated by treating the underlying disease. For hyperpigmented lesions, hydroquinone may be used in cases that enhance with Wood’s light. Tretinoin application may enhance the effect of hydroquinone. Laser treatments and chemical peels must be done with extreme caution, because results are unpredictable and increased pigmentation may result. In darker patients, especially with lichenoid diseases such as lichen planus, the dyspigmentation can last for years.
Abad-Casintahan F, et al: Frequency and characteristics of acne-related post-inflammatory hyperpigmentation. J Dermatol 2016; 43: 826.
Al Mohizea S: The effect of menstrual cycle on laser-induced hyperpigmentation. J Drugs Dermatol 2013; 12: 1335.
Grimes PE: Management of hyperpigmentation in darker racial ethnic groups. Semin Cutan Med Surg 2009; 28: 77.
Kasuya Y, et al: Glossal pigmentation caused by the simultaneous uptake of iron and tea. Eur J Dermatol 2014; 24: 493.
Lobo C, et al: Retrospective epidemiological study of Latin American patients with transfusional hemosiderosis. Hematology 2011; 16: 265.
Oram Y, Akkaya AD: Refractory postinflammatory hyperpigmentation treated with fractional CO 2 laser. J Clin Aesthet Dermatol 2014; 7: 42.
Payette MJ, et al: Lichen planus and other lichenoid dermatoses. Clin Dermatol 2015; 33: 631.
Thompson J, et al: Severe haemosiderin pigmentation after intravenous iron infusion. Intern Med J 2014; 44: 706.
Melasma (Chloasma)
Melasma is a common disorder, with two predisposing factors: sun exposure and sex hormones. It tends to affect darker-complexioned individuals, especially East, West, and Southeast Asians, Hispanics, and black persons who live in areas of intense sun exposure and who have Fitzpatrick skin types IV and V. Subtle melasma, as identified by UV light examination, may be seen in up to 30% of middle-aged Asian females. Men are also affected, especially those from Central America.
The pathogenesis of melasma is not known. However, many observations strongly suggest that sun exposure is the primary trigger. Melasma affects the face, a sun-exposed area, and worsens in the summer. Melasma patients have a lower minimal erythema dose (MED) to UV light, and pigment more easily with UV exposure. An association exists between the number of melanocytic nevi and the development of melasma. The prevalence of melasma increases with age in both men and women. Solar elastosis is more marked in areas of melasma than unaffected facial skin. Melasma-affected skin has reduced WIF-1 (Wnt antagonist) expression and resultant increased Wnt expression; Wnt stimulates melanogenesis.
After sun exposure, the second most important trigger for melasma is female hormones. Melasma is more common and severe in women than men. It occurs frequently during pregnancy, with oral contraceptive (OC) use, or with hormone replacement therapy (HRT) at menopause. Discontinuing OC use or HRT rarely clears the pigmentation, which still may last for many years. In contrast, melasma of pregnancy usually clears within a few months of delivery. Melasma may be seen in other endocrinologic disorders, as well as with phenytoin and finasteride therapy.
Melasma is characterized by brown patches, typically on the malar prominences and forehead. The forearms may also be affected. There are three clinical patterns of facial melasma: centrofacial, malar, and mandibular. The centrofacial and malar patterns constitute the majority ( Fig. 36.3 ), but most patients have multiple types, so this classification is not very useful therapeutically. The pigmented patches are usually sharply demarcated. Although melasma has classically been classified as epidermal or dermal, based on the presence or absence of Wood’s light enhancement, respectively, most cases show both epidermal and dermal melanin. There are nuclear changes within keratinocytes that indicate that epidermal melanin units are altered. Dermal melanophages are a normal finding in sun-exposed Asian skin. Independent of Wood’s light findings, a therapeutic trial of some form of hypopigmenting agent should be offered.
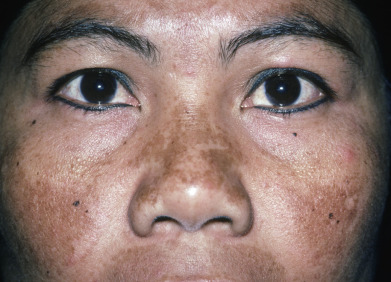
Therapeutically, a sunblock with broad-spectrum UVA (even visible light) coverage should be used daily; it will modestly improve the melasma, but more important, will enhance the efficacy of bleaching creams and help prevent new lesions. Bleaching creams with hydroquinone are the gold standard and are moderately efficacious, containing 2% (available over the counter) to 4% hydroquinone. Tretinoin cream may be added to increase efficacy. Tretinoin alone may reduce melasma, but it is not as effective as hydroquinone. The combination of hydroquinone and tretinoin, administered with a topical corticosteroid, has been called “Kligman’s formula” and is the most effective topical regimen available to treat melasma. Twice-weekly application of the triple combination can be effective for maintenance. Overuse can lead to fixed erythema and telangiectasias, acneiform eruptions, and hypertrichosis. Overuse of hydroquinone can lead to exogenous ochronosis. When 4% hydroquinone is ineffective, higher concentrations may be recommended. Satellite pigmentation and local ochronosis are potential complications from use of these higher-concentration preparations. Methimazole, azelaic acid, kojic acid, vitamin C, and arbutin are other therapies with minimal to moderate efficacy. Many of these agents are added to cosmetic products for skin lightening and may be combined, because they act on different steps of melanogenesis. All these topical agents are generally less effective than 4% hydroquinone but may be used in the patient intolerant of hydroquinone. Oral tranexamic acid may play a role as a systemic agent in treating refractory melasma.
Various surgical procedures, such as peels and light-based treatments, have been proposed as effective for melasma, but results are mixed. Peels with glycolic acid, salicylic acid, trichloroacetic acid (TCA), and tretinoin 1% have not reproducibly enhanced the efficacy of 4% hydroquinone and can cause hyperpigmentation if irritation ensues. The use of light-based modalities for the treatment of melasma should be approached with caution. These therapies may be complicated by hyperpigmentation, irritation, hypopigmentation, and even scarring, if not used appropriately. Intense pulse light (IPL) can improve melasma, but there is a high relapse rate. Pulsed dye laser may enhance combination topical treatment, and improvement may continue after therapy is discontinued. Q-switched neodymium:yttrium-aluminum-garnet (Nd:YAG) laser therapy can lead to increased pigmentation.
Adalatkhah H, et al: Melasma and its association with different types of nevi in women. BMC Dermatol 2008; 8: 3.
Brianezi G, et al: Changes in nuclear morphology and chromatin texture of basal keratinocytes in melasma. J Eur Acad Dermatol Venereol 2015; 29: 809.
Castanedo-Cazares JP, et al: Near-visible light and UV photoprotection in the treatment of melasma. Photodermatol Photoimmunol Photomed 2014; 30: 35.
Córdova ME, et al: Exogenous ochronosis in facial melasma. Actas Dermosifiliogr 2017; 108: 381.
Famenini S, et al: Finasteride-associated melasma in a Caucasian male. J Drugs Dermatol 2014; 13: 484.
Fisk WA, et al: The use of botanically derived agents for hyperpigmentation. J Am Acad Dermatol 2014; 70: 352.
Handel AC, et al: Melasma. An Bras Dermatol 2014; 89: 771.
Handel AC, et al: Risk factors for facial melasma in women. Br J Dermatol 2014; 171: 588.
Hernandez-Barrera R, et al: Solar elastosis and presence of mast cells as key features in the pathogenesis of melasma. Clin Exp Dermatol 2008; 33: 305.
Hexsel D, et al: Epidemiology of melasma in Brazilian patients. Int J Dermatol 2014; 53: 440.
Hexsel D, et al: Objective assessment of erythema and pigmentation of melasma lesions and surrounding areas on long-term management regimens with triple combination. J Drugs Dermatol 2014; 13: 444.
Jutley GS, et al: Systematic review of randomized controlled trials on interventions for melasma. J Am Acad Dermatol 2014; 70: 369.
Kandhari R, Khunger N: Skin lightening agents. Indian J Dermatol Venereol Leprol 2013; 79: 701.
Kim MJ, et al: Punctate leucoderma after melasma treatment using 1064-nm Q-switched Nd:YAG laser with low pulse energy. J Eur Acad Dermatol Venereol 2009; 23: 960.
Kim NH, et al: Cadherin 11, a miR-675 target, induces N-cadherin expression and epithelial-mesenchymal transition in melasma. J Invest Dermatol 2014; 134: 2967.
Li Y, et al: Treatment of melasma with oral administration of compound tranexamic acid. J Eur Acad Dermatol Venereol 2014; 28: 388.
Malek J, et al: Successful treatment of hydroquinone-resistant melasma using topical methimazole. Dermatol Ther 2013; 26: 69.
Passeron T: Long-lasting effect of vascular targeted therapy of melasma. J Am Acad Dermatol 2013; 69: e141.
Rivas S, Pandya AG: Treatment of melasma with topical agents, peels and lasers. Am J Clin Dermatol 2013; 14: 359.
Sardana K, Garg VK: Lasers are not effective for melasma in darkly pigmented skin. J Cutan Aesthet Surg 2014; 7: 57.
Sardana K, et al: Which therapy works for melasma in pigmented skin. Indian J Dermatol Venereol Leprol 2013; 79: 420.
Trivedi MK, et al: A review of laser and light therapy in melasma. Int J Womens Dermatol 2017; 3: 11.
Tse TW, et al: Tranexamic acid. J Cosmet Dermatol 2012; 12: 57.
Reticulate Pigment Disorders of the Skin
This group of disorders is linked by similar clinical features: reticulate pigmentation of various skin sites and characteristic histology—adenoid pigmented proliferations of the rete ridges of the interfollicular and infundibular follicular epidermis, at times with focal acantholysis. Patients may have overlapping features of several different syndromes, but now that the genetic basis for disease is being discovered, correct categorization should be easier.
Dyschromatosis Symmetrica Hereditaria (Reticulate Acropigmentation of Dohi)
Originally described and still reported primarily in the Japanese, acropigmentation of Dohi has been found to affect individuals from Europe, India, and the Caribbean region. It is also referred to as dyschromatosis symmetrica hereditaria (DSH) or symmetric dyschromatosis of the extremities. It is inherited most often as an autosomal dominant trait, although autosomal recessive kindreds have been reported. Patients develop progressive hyperpigmented and hypopigmented macules, often mixed in a reticulate pattern, concentrated on the dorsal extremities, especially the dorsal hands and feet. The lesions vary in size from pinpoint to pea sized. Freckle-like macules can present on the face. Long hair on the forearms, hypopigmented or hyperpigmented hair, acral hypertrophy, and dental abnormalities also have been reported. Lesions appear in infancy or early childhood and usually stop spreading before adolescence. The pigmentary lesions last for life although there is a report of spontaneous resolution. The autosomal dominant form of DSH is caused by a mutation in the DSRAD (ADAR1) gene.
Kantaputra PN, et al: Dyschromatosis symmetrica hereditaria with long hair on the forearms, hypo/hyperpigmented hair, and dental anomalies. Am J Med Genet A 2012; 158: 2258.
Mohana D, et al: Reticulate acropigmentation of Dohi. Indian J Dermatol 2012; 57: 42.
Murata T, et al: Dyschromatosis symmetrica hereditaria with acra hypertrophy. Eur J Dermatol 2011; 21: 649.
Shi BJ, et al: First report of the coexistence of dyschromatosis symmetrica hereditaria and psoriasis. J Eur Acad Dermatol Venereol 2012; 26: 657.
Zhang SD, et al: Pathogenicity of ADAR1 mutation in a Chinese family with dyschromatosis symmetrica hereditaria. J Eur Acad Dermatol Venereol 2017; 31: e483.
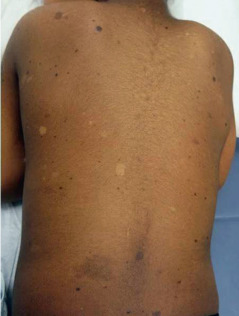
Dyschromatosis Universalis Hereditaria, Familial Progressive Hyperpigmentation and Hypopigmentation
Dyschromatosis universalis hereditaria (DUH) is a rare autosomal dominant genodermatosis characterized by asymptomatic hyperpigmented and hypopigmented macules ( Fig. 36.4 ) in a generalized distribution on the trunk and limbs, or sometimes the face. Lesions are irregular in size and shape and appear in infancy or childhood, often in the first few months of life. The palms, soles, and mucous membranes are usually spared. Most DUH patients do not show other symptoms and are otherwise well. Infrequently reported associations include ocular and auditory abnormalities, photosensitivity, developmental delay, and short stature. Histologically, there are normal numbers of melanocytes in both the lighter and the darker skin, but more melanized, mature melanosomes in the darker areas and empty, immature melanosomes in the hypopigmented areas. A mutation in the ABCB6 gene (mitochondrial porphyrin transporter localized to outer membrane of mitochondria) has been identified in numerous cases of autosomal dominant DUH (DUH-1). The wild-type protein localizes to the dendrites of melanocytes and is probably involved in melanosome transport. The mutant protein remains in the Golgi complex, which could disrupt melanosome transport.
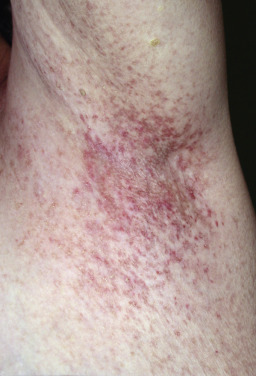
A much rarer variant of DUH is DUH-2. It is inherited as an autosomal recessive genodermatosis, with a putative gene location on chromosome 12.
Familial progressive hyperpigmentation (FPH) is an autosomal dominant genodermatosis characterized by hyperpigmented patches presenting in early infancy and progressing with age. Hypopigmented lesions are absent, distinguishing it from DUH-2, which FPH otherwise closely resembles. Familial progressive hyperpigmentation and hypopigmentation (FPHH) is an autosomal dominant disorder characterized by diffuse, partly blotchy hyperpigmentation, hyperpigmented macules, café au lait macules, and larger hypopigmented ash-leaf macules on the face, neck, trunk, and limbs present at birth or early in infancy ( Fig. 36.5 ). Lesions increase in size and number with age. FPHH and FPH have also been associated with mutations in the same region of chromosome 12 as DUH-2, mapping to the gene KITLG (also known as steel factor or mast cell growth factor/stem cell factor), which codes for the ligand of c-KIT. The mutations in FPHH and FPH are gain-of-function mutations. c-KIT mutations are also implicated in mastocytomas and there is a report of a patient with both FPH and mastocytomas.
Amyere M, et al: KITLG mutations cause familial progressive hyper- and hypopigmentation. J Invest Dermatol 2011; 1331: 1234.
Cui YX, et al: Novel mutations of ABCB6 associated with autosomal dominant dyschromatosis universalis hereditaria. PLoS One 2013; 8: e79808.
Nogita T, et al: Removal of facial and labial lentigines in dyschromatosis universalis hereditaria with a Q-switched alexandrite laser. J Am Acad Dermatol 2011; 65: e61.
Piqueres-Zubiaurre T, et al: Familial progressive hyperpigmentation, cutaneous mastocytosis, and gastrointestinal stromal tumor as clinical manifestations of mutations in the c-KIT receptor gene. Pediatr Dermatol 2017; 34: 84.
Sorensen RH, et al: Dyschromatosis universalis hereditaria with oral leukokeratosis. Pediatr Dermatol 2015; 32: e283.
Zhang C, et al: Mutations in ABCB6 cause dyschromatosis universalis hereditaria. J Invest Dermatol 2013; 133: 2221.
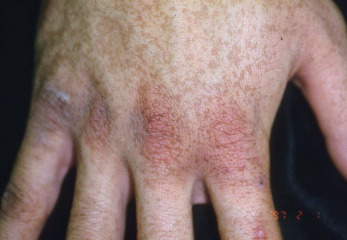
Dowlin-Degos Disease (Reticular Pigmented Anomaly of Flexures)
Reticular pigmented anomaly of the flexures is a rare autosomal dominant pigmentary disorder; it is now more often called Dowling-Degos disease (DDD). Pigmentation usually appears at puberty or in early adolescence but may present later in adulthood. The skin lesions primarily affect the intertriginous areas, such as the axillae, neck, genitalia, and inframammary/sternal areas. In some cases, the dorsal hands are involved. The pigmentation is reticular; at the periphery, discrete, brownish black macules surround the partly confluent, central pigmented area and progresses very slowly. In more mildly affected patients, the pigmentation is dappled. A follicular variant of DDD has been reported. There are frequently acneiform, pitted scars, sometimes pigmented, around the mouth. Comedonal and cystic lesions have been described on the flexures and in the axillae. Hidradenitis suppurativa–like lesions in the groin and axilla may occur. Patients may complain that the condition is worse during hot weather. Squamous cell carcinoma of the buttocks or perianal area has been described.
Histologically, in addition to the typical lentiginous adenoid proliferations of the rete ridges, small horn cysts may be present, so that the pattern resembles that of a reticulated seborrheic keratosis. Comedones may be present. Classic autosomal dominant DDD is caused by mutations in the keratin 5 gene (KRT5). Similar mutations occur in Galli-Galli disease, suggesting that the two conditions represent variants of the same disorder rather than separate diseases. In DDD patients who lack mutation in KRT5, mutations have been found in POGLUT1 and POFUT1, both of which are essential regulators of Notch activity.
Galli-Galli Disease
Galli-Galli disease is now recognized as an acantholytic variant of DDD, also caused by mutations in the KRT5 gene. The skin lesions are 1–2 mm, slightly keratotic, red to dark-brown papules, which are focally confluent in a reticulate pattern ( Fig. 36.6 ). The skin lesions favor skinfolds, although other skin sites may also be involved. The neck, axillae, upper extremities, dorsal hands, trunk, groin, and even the scrotum and lower extremities may be affected. Histologically, there is prominent digitate downgrowth of the rete ridges, identical to that seen in DDD. The characteristic histologic feature is a suprabasilar cleft and suprapapillary thinning of the epidermis. There is no dyskeratosis, as seen in Grover disease. Ablative laser treatment led to axillary symptom resolution in one patient.
Basmanav FB, et al: Mutations on POGLUT1, encoding protein O-glucosyltransferase 1, cause autosomal-dominant Dowling-Degos disease. Am J Hum Genet 2014; 94: 135.
Gomes J, et al: Galli-Galli disease. Case Rep Med 2011; 2011: 703257.
Hanneken S, et al: Systematic mutation screening of KRTS supports the hypothesis that Galli-Galli disease is a variant of Dowling-Degos disease. Br J Dermatol 2010; 163: 197.
Horner ME, et al: Dowling-Degos disease involving the vulva and back. Dermatol Online J 2011; 17: 1.
Muller CS, et al: The spectrum of reticulate pigment disorders of the skin revisited. Eur J Dermatol 2012; 22: 596.
Schmieder A, et al: Galli-Galli disease is an acantholytic variant of Dowling-Degos disease. J Am Acad Dermatol 2012; 66: e250.
Singh S, et al: Follicular Dowling-Degos disease. Indian J Dermatol Venereol Leprol 2013; 79: 802.
Taskapan O, et al: Dowling-Degos disease with diffuse penile pigmentation. J Eur Acad Dermatol Venereol 2014; 28: 1405.
Voth H, et al: Efficacy of ablative laser treatment in Galli-Galli disease. Arch Dermatol 2011; 147: 317.
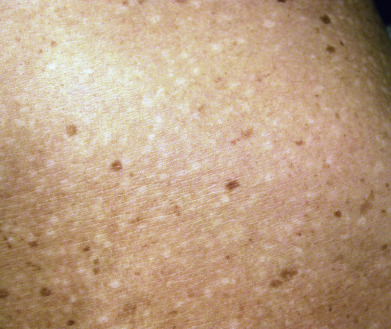
Reticulate Acropigmentation of Kitamura
Reticulate acropigmentation of Kitamura (RPK) is a rare autosomal dominant disease that initially was recognized in Japan but now has been seen in many countries. The characteristic presentation is pigmented, angulated, irregular, freckle-like lesions with atrophy ( Fig. 36.7 ), arranged in a reticulate pattern on the dorsal feet and hands. Lesions start in the first to second decade of life, gradually progress, and slowly darken over time. The axillae and groin may be affected, as can the skin of the trunk and more proximal extremities. Linear irregular breaks in the dermatoglyphics of the palms are characteristic and help to distinguish this disorder from the other “reticulate flexural anomalies.” Patients with mixed features of DDD and RPK have been reported. RPK is caused by a loss-of-function mutation in ADAM10, which also affects the NOTCH pathway explaining the similarities with DDD.
Koguchi H, et al: Characteristic findings of handprint and dermoscopy in reticulate acropigmentation of Kitamura. Clin Exp Dermatol 2014; 39: 58.
Ralser DJ, et al: Functional implications of novel ADAM10 mutations in reticulate acropigmentation of Kitamura. Br J Dermatol 2017; 177: e340.
Dermatopathia Pigmentosa Reticularis
Dermatopathia pigmentosa reticularis (DPR) is an extremely rare dominant ectodermal dysplasia characterized by the triad of generalized reticulate hyperpigmentation, noncicatricial alopecia, and onychodystrophy. Additional associations include loss of dermatoglyphics, hypohidrosis or hyperhidrosis, pigmented lesions of the oral mucosa, palmoplantar hyperkeratosis, and nonscarring blisters on the dorsa of the hands and feet. Wiry scalp hair and digital fibromatosis have also been reported. Both Naegeli-Franceschetti-Jadassohn syndrome (NFJS) and DPR are caused by mutations in the keratin 14 gene. Patients with NFJS can be differentiated from DPR because NFJS patients have dental anomalies their pigmentation may fade.
Al Saif F: Dermatopathia pigmentosa reticularis. Indian J Dermatol 2016; 61: 468.
Goh BK, et al: A case of dermatopathia pigmentosa reticularis with wiry scalp hair and digital fibromatosis resulting from a recurrent KRT14 mutation. Clin Exp Dermatol 2009; 34: 340.
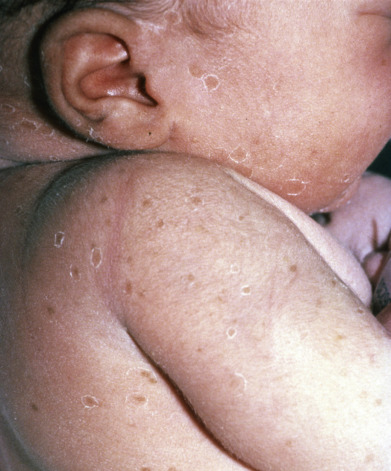
Transient Neonatal Pustular Melanosis
Also called transient pustular melanosis (TPM) is present at birth. Newborns present with 1–3 mm, flaccid, superficial fragile pustules. Some of the pustules may have already resolved in utero, leaving pigmented macules ( Fig. 36.8 ). Lesions can occur anywhere. Numerous lesions and lesions up to 1 cm in diameter have been reported, but any new pustules forming after birth should raise suspicion for another benign pustulosis such as erythema toxicum neonatorum or, more important, infections such as Staphylococcus aureus or herpes simplex. In dark-skinned infants, pigmented macules may persist for weeks or months after the pustules have healed. Transient neonatal pustular melanosis is observed more frequently in infants with skin types III-VI (approximately 5% vs less than 1% of infants with skin types I-II) and may be more common after vaginal than cesarean delivery.
Histologically, there are intracorneal or subcorneal aggregates, predominantly of neutrophils, although eosinophils may also be found. Dermal inflammation is composed of a mix of neutrophils and eosinophils.
Brazzelli V, et al: An unusual case of transient neonatal pustular melanosis. Eur J Pediatr 2014; 173: 1655.
Ekiz O, et al: Skin findings in newborns and their relationship with maternal factors. Ann Dermatol 2013; 25: 1.
Paloni G, Cutrone M: Giant transient pustular melanosis in an infant. Arch Dis Child Fetal Neonatal Ed 2013; 98: F492.
Peutz-Jeghers Syndrome
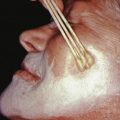
Peutz-Jeghers syndrome (PJS) is characterized by hyperpigmented macules on the lips and oral mucosa, polyposis of the gastrointestinal (GI) tract, and greatly increased cancer risk. The dark-brown or black macules appear typically on the lips, especially the lower lip, in infancy or early childhood ( Fig. 36.9 ). Similar lesions may appear on the buccal mucosa, tongue, gingiva, and the perianal mucosa; macules may also occur around the mouth, on the central face, perianally, and on the backs of the hands, especially the fingers, toes, and tops of the feet. More than two thirds of patients have lesions on the hands and feet, and 95% have perioral lesions. Skin lesions grow in size and number until puberty, after which they begin to regress. Buccal pigmented macules tend to persist. Similar pigmentation may be seen in the bowel.
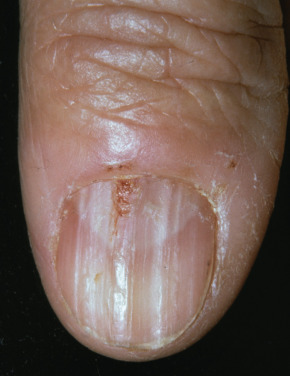
The diagnosis of PJS is made with any of four major criteria: (1) two or more histologically confirmed PJS polyps; (2) any number of PJS polyps and a family history of PJS; (3) characteristic mucocutaneous pigmentation and a family history of PJS; or (4) any number of PJS polyps and characteristic mucocutaneous pigmentation. In 94% of patients who fulfill these criteria, a mutation in the STK11 gene will be found. Almost half of patients are de novo mutations.
The associated polyps, which are histologically characteristic, are most common in the small intestine, but may also occur in the stomach, colon, and least commonly the rectum. The polyposis of the small intestine may cause repeated bouts of abdominal pain and vomiting. Bleeding and intussusception are common; intussusception is frequent (47%). Boys with PJS often have evidence of estrogen excess with gynecomastia and advanced bone age.
Patients with PJS have a 10-fold to 18-fold greater lifetime cancer risk (81%–94%) than the general population. The greatest risk is for GI malignancy, which is increased 130-fold in PJS patients. These cancers occur in the colon (39% of patients), stomach (29%), and small intestine (13%). Cancers begin to appear about age 30 years. Cancers also occur in extraintestinal sites, especially the breast, genitourinary (GU) tract, and pancreas (100-fold increase in PJS patients). The prevalence of cancers by anatomic site is pancreas (26%), breast (54%, can be bilateral), and ovary (21%). Sertoli-Leydig cell stromal tumors occur in 9% of PJS males, and sex cord tumors with annular tubules can occur in female PJS patients. Given the high risk of cancer in PJS patients, standard screening protocols have been recommended. Because 40% of patients develop significant GI and potentially GU complications by age 6 years, GI screening may need to begin as early as age 4 or 5, with testicular examination in males with PJS. The syndrome is caused by a germline mutation of the STK11/LKB1 tumor suppressor gene. Patients with truncation of the gene rather than a missense mutation are more severely affected, suggesting a phenotype/genotype correlation. In Chinese PJS patients, mutations in OR4C45, ZAN, pre–micro-RNAs, and other genes have been identified, suggesting that multiple different genes can cause this syndrome.
Laugier-Hunziker syndrome, Carney syndrome, and Cronkhite-Canada syndrome should be considered in the differential diagnosis of PJS. Laugier-Hunziker syndrome presents with mucosal pigmentation and pigmented nail streaks. Cronkhite-Canada syndrome consists of melanotic macules on the fingers and GI polyposis, as well as generalized, uniform darkening of the skin, extensive alopecia, and onychodystrophy. The polyps that occur are usually benign adenomas and may involve the entire GI tract. A protein-losing enteropathy may develop and is associated with the degree of intestinal polyposis. Onset is typically after age 30 in this sporadically occurring, generally benign condition. Hypogeusia (reduced taste) is the dominant initial symptom in Cronkhite-Canada, followed by diarrhea and ectodermal changes. The majority of all cases have been reported from Japan. Zinc therapy may improve the hypogeusia and other symptoms. Carney syndrome patients may also develop Sertoli cell tumors and gynecomastia, which, in combination with their mucocutaneous pigmentation, may lead to an erroneous diagnosis of PJS.
Banno K, et al: Hereditary gynecological tumors associated with Peutz-Jeghers syndrome (review). Oncol Lett 2013; 6: 1184.
Campos-Munoz L, et al: Dermoscopy of Peutz-Jeghers syndrome. J Eur Acad Dermatol Venereol 2009; 23: 730.
Goldstein SA, Hoffenberg EJ: Peutz-Jegher syndrome in childhood. J Pediatr Gastroenterol Nutr 2013; 56: 191.
Ham S, et al: Overexpression of aromatase associated with loss of heterozygosity of the STK11 gene accounts for prepubertal gynecomastia in boys with Peutz-Jeghers syndrome. J Clin Endocrinol Metab 2013; 98: E1979.
Jelsig AM, et al: Hamartomatous polyposis syndromes. Orphanet J Rare Dis 2014; 9: 101.
Korsse SE, et al: Pancreatic cancer risk in Peutz-Jeghers syndrome patients. J Med Genet 2013; 50: 59.
Resta N, et al: Cancer risk associated with STK11/LKB1 germline mutations in Peutz-Jeghers syndrome patients. Dig Liv Dis 2013; 45: 606.
Richey JD, et al: Carney syndrome in a patient previously considered to have Peutz-Jeghers syndrome. J Am Acad Dermatol 2014; 70: e44.
Shah KR, et al: Cutaneous manifestations of gastrointestinal disease. Part I. J Am Acad Dermatol 2013; 68: 189.e1.
Tsai HL, et al: Rectal carcinoma in a young female patient with Peutz-Jeghers syndrome. Med Princ Pract 2014; 23: 89.
Wang HH, et al: Exome sequencing revealed novel germline mutations in Chinese Peutz-Jeghers syndrome patients. Dig Dis Sci 2014; 59: 64.
Wangler MF, et al: Unusually early presentation of small-bowel adenocarcinoma in a patient with Peutz-Jeghers syndrome. J Pediatr Hematol Oncol 2013; 35: 323.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


