Hair
Hair is a protein by-product of follicles distributed everywhere on the body surface except the palms, soles, vermilion portion of the lips, glans penis, penile shaft, nailbeds, and sides of the fingers and toes. Although hair is of minimal functional benefit to humans, the psychologic effects of disturbances of hair growth are commonly a source of great concern to children, adolescents, and their parents.
In the human fetus, groups of cells appear in the epidermis at about the eighth week of gestation. These differentiate to form the hair follicles, and hair begins to develop between the eighth and twelfth weeks of fetal life. This growth continues throughout fetal development. Although there are indications that some hair is lost during gestation and at the time of birth, the majority of hairs on the newborn are 5 to 6 months old.
Lanugo hairs are fine, soft, unmedullated, and poorly pigmented hairs seen only in fetal and neonatal life, except in the rare hereditary syndrome hypertrichosis lanuginosa. They appear as a fine, dense growth over the entire cutaneous surface of the fetus. Lanugo hair is normally shed in utero in the seventh or eighth month of gestation but may cover the entire cutaneous surface of the premature newborn infant. Postnatal hair may be divided into vellus and terminal types. Vellus hairs are the fine, lightly pigmented hairs seen on the arms and faces of children. Terminal hairs are the mature, thick, darker hairs on the scalp, eyebrows, eyelashes, and areas of secondary-sexual hair distribution. The number and distribution of individual hair follicles are genetically determined and constant from birth. As the infant’s skin grows, however, the density of hair follicles reduces from 1135 per cm 2 at birth to 615 per cm 2 by adulthood.
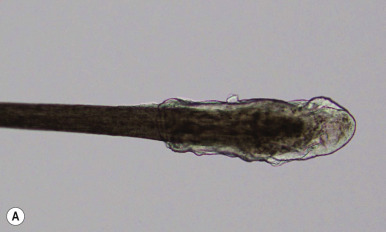
The average human scalp contains 100,000 hairs. The average growth rate of terminal hair is approximately 2.5 mm/week (1 cm/month). The hair shaft represents the equivalent of the stratum corneum of skin, with the follicular keratinocytes dictating the characteristics of the shaft. The hair root is characterized by three definable cyclic stages of growth: anagen ( Fig. 7-1, A ), catagen, and telogen ( Fig. 7-1, B ; Box 7-1 ). The human hair follicle has a fairly long phase of regular growth (the anagen phase) that lasts 2 to 6 years, with an average of 3 years. The hairs then undergo a period of partial degeneration (the catagen phase), lasting up to 3 weeks, followed by a resting (telogen or club) phase. The telogen phase of the follicle lasts for about 3 months. At the end of this time, new growth is initiated. As new hairs grow, they push out the old telogen hairs that have remained in the resting follicles. In healthy individuals, 85% to 90% of the scalp is in the actively growing anagen stage, and 1% is in the brief transitional (catagen) stage; 10% to 15% is in the resting or telogen stage, with an average of 50 to 100 hairs shed and simultaneously replaced each day. Although scalp hair has a long anagen phase, eyelash and extremity hair have a lower anagen/telogen ratio and thus tend to be shorter.

- 1.
Anagen phase (active growth phase) lasts 2 to 6 years (average 3 years)
- 2.
Catagen phase (stage of partial degeneration) lasts 10 to 14 days
- 3.
Telogen phase (resting stage) lasts 3 to 4 months
Neonatal Hair
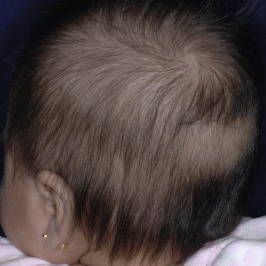
The first crop of terminal scalp hair is in the actively growing anagen phase at birth, but within the first few days of life there is a physiologic conversion to the telogen phase. Consequently a high proportion of neonatal scalp hairs are shed during the first 4 months of life ( Fig. 7-2 ). This telogen shedding (telogen effluvium of the newborn) usually is gradual hair loss, particularly between 2 and 4 months and most noticeable at the occipital area. Rarely the hair loss occurs in the first 4 weeks of life in a frontal-temporal pattern. This physiologic hair shedding at the occipital area is not related to the baby’s sleeping position (i.e., friction). Replacement of the first terminal hairs is generally completed before the first 6 months of life. The neonatal hairline commonly extends along the forehead and temples to the lateral margin of the eyebrows. These terminal hairs gradually convert to vellus hairs during the first year of life. Premature infants are often covered by lanugo hairs, which are more densely distributed on the face, limbs, and trunk. This retention of lanugo hair is probably related to the cyclic activity in utero and the normal shedding of telogen vellus hairs in the fetus during the last few weeks of gestation.
Alopecias
Clinical examination allows hair loss disorders to be divided into nonscarring (noncicatricial) or scarring (cicatricial) types ( Box 7-2 ). Causes of nonscarring alopecia include alteration of the hair growth cycle, inflammatory cutaneous disease, and structural abnormalities of the hair. Some traumatic disorders such as traction alopecia, pressure alopecia, or trichotillomania can scar if severe but often resolve without clinical evidence of scarring. Evaluation typically involves gentle traction on the hair (hair-pull test) to determine if hair comes out easily (as in loose anagen syndrome, alopecia areata [AA], or telogen effluvium) and microscopic or trichoscopic evaluation of hair shafts to seek hair shaft abnormalities ( Box 7-3 ).
- 1.
Overall characteristics: pattern of loss, extent, color, texture, length, breakage
- 2.
Scalp features: scarring vs. nonscarring, inflammation, trichoscopy (follicular openings), pigmentary change, scaling, crusts, pustules
- 3.
Hair beyond scalp: eyebrows, lashes, extremity hair, secondary sexual hair if adolescent
- 4.
Special tests: trichoscopy of shaft, hair pull, hair tug, hair mount (see Box 7-3)
Hair pull : Grasp about 50 hairs between first three fingers and pull gently but firmly away from scalp at four scalp regions (i.e., frontal, occipital, temporal bilaterally); positive pull test means more than 10% of hairs pull out easily (are in telogen) and is seen with active alopecia areata.
Hair tug : Hold a group of hairs with one hand and use other hand to pull away at distal end, looking for hair breakage (which can then be analyzed microscopically in a hair mount). This can be helpful with fragile hair syndromes such as monilethrix.
Hair mount : Hairs are separated, oriented side-by-side on a glass slide, covered with mounting medium (such as Permount), and covered with a coverslip without air bubbles. This technique allows one to distinguish anagen from telogen bulbs based on shape and pigmentation, distorted bulbs and cuticles (as in loose anagen syndrome), and hair shaft abnormalities.
Trichoscopy : Can replace hair mounts and microscopy when examining shaft. Disease-specific findings such as for alopecia areata (follicular openings showing yellow dots; “exclamation-point” hairs), tinea capitis (comma or corkscrew hairs), and trichotillomania (coiled or flame hairs); black dots at follicular opening suggest broken hairs and can be seen with alopecia areata, tinea, and trichotillomania.
Nonscarring Alopecias with Hair Shaft Abnormalities
Hair Shaft Abnormalities with Increased Fragility
Variations in the structure of the hair shaft are a common occurrence and at times may provide clues to other pathologic abnormalities ( Box 7-4 ). Because each hair shaft anomaly has a distinctive morphology, the diagnosis often can be established in the office by trichoscopy (using a dermatoscope) (see Box 7-3 ) or by microscopic examination of snipped hairs. Other than reduction of trauma to reduce breakage, there is no effective treatment for this group of disorders.
Trichorrhexis Nodosa
- ▪
Trauma (most common)
- ▪
Argininosuccinic aciduria
- ▪
Citrullinemia
- ▪
Oculodentodigital dysplasia
- ▪
Trichothiodystrophy
- ▪
Trichohepatoenteric syndrome
- ▪
Monilethrix
- ▪
Hair keratin mutations (KRT81, KRT83, KRT86)
- ▪
Desmoglein 4 mutations
- ▪
Trichorrhexis Invaginata
- ▪
Netherton syndrome
- ▪
Pili Torti
- ▪
Menkes syndrome
- ▪
Crandall syndrome
- ▪
Björnstad syndrome
- ▪
Bazek–Dupré–Christol syndrome
- ▪
Rombo syndrome
- ▪
Hypotrichosis with juvenile macular degeneration
- ▪
Mitochondrial enzyme defects
- ▪
Trichothiodystrophy
- ▪
Group I: mutations in XPD, XPB, p8
- ▪
Group II: mutations in TTDN1
- ▪
Group III: Pollitt syndrome, Sabinas syndrome (genes unknown)
- ▪
Trichorrhexis Nodosa.
Trichorrhexis nodosa, the most common hair shaft anomaly, is a distinctive disorder manifested by increased fragility. Grayish-white nodules may be seen on the hair ( Fig. 7-3 ), which under trichoscopy or light microscopy have the appearance of two interlocking brushes or brooms, the result of segmental longitudinal splitting of fibers without complete fracture. The disorder features dry, lusterless, short hair that is easily fractured.

Usually “acquired” in adolescents without other issues, trichorrhexis nodosa most commonly results from trauma to the hair. The injury may result from the use of hot combs, excessively hot hairdryers, hair straighteners, other chemical treatments, or from the cumulative cuticular damage from vigorous combing and brushing, repeated salt-water bathing, prolonged sun exposure, and frequent shampooing. Cream rinses and protein conditioners are helpful. If hair-straightening procedures, vigorous grooming habits, and thermal and chemical trauma to the hair are discontinued, the acquired form of trichorrhexis nodosa generally improves within 2 to 4 years.
Less commonly trichorrhexis nodosa is genetic and manifests during infancy. Infants with the autosomal dominant form show normal hair at birth, but the hair that regrows within a few months is abnormal; the hair defect tends to improve with advancing age. Trichorrhexis nodosa may also be a manifestation of children with the late-onset form of argininosuccinic aciduria, a condition that results from lack of argininosuccinase. In this condition, the hair is usually normal at birth and first becomes fragile at 1 to 2 years of age with a dull, matted appearance, especially at the occipital area. The hair defects are associated with psychomotor retardation, cerebellar ataxia, and a marked increase of argininosuccinic acid in the blood, urine, and cerebrospinal fluid. Dietary treatment of the metabolic abnormality leads to normalization of the appearance and integrity of the hair. Similar clinical manifestations are found in infants with citrullinemia, caused by a deficiency of argininosuccinic acid synthetase. Trichorrhexis nodosa is the most common hair shaft abnormality, but pili torti has been described and hair bulbs may be atrophic. Some patients show an eruption that resembles acrodermatitis enteropathica. Trichorrhexis nodosa has also been described in oculodentodigital dysplasia (see Group 2 Ectodermal Dysplasia section), trichothiodystrophy (TTD), and trichohepatoenteric syndrome, characterized by facial dysmorphism, liver disease, immune defects, and severe diarrhea requiring intravenous nutrition.
Monilethrix.
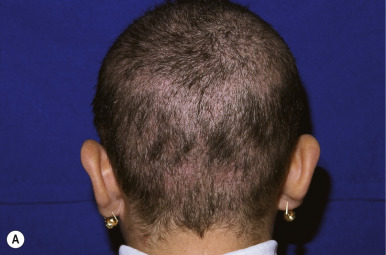
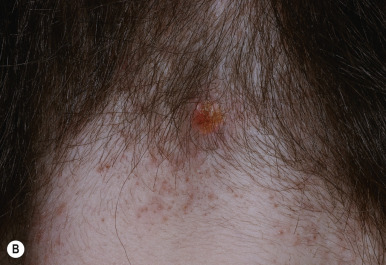
Monilethrix (beaded hair) is an autosomal dominant disorder characterized by partial alopecia from breakage and variation in hair shaft thickness with small node-like deformities that produce a beaded appearance and internodal fragility. The nodal pattern can be seen by trichoscopy but can be subtle. In individuals with this disorder, normal neonatal lanugo hairs are shed during the first few weeks of life. The regrown hair, which generally appears at about the second month of life, is dry, lusterless, and brittle and fails to grow to any appreciable length because of breakage ( Fig. 7-4, A ). In severe cases, the infant may remain bald or the scalp hair may be sparse, easily fractured, and stubble-like with follicular prominence from keratosis. Although generally a disorder of scalp hair, body hairs may also be affected. The clinical findings are limited to the occiput and nape in more limited cases ( Fig. 7-4, B ). Occasionally this disorder is not apparent during infancy but becomes apparent later in childhood or during adult life. Follicular keratosis is associated in some pedigrees and may affect the face, scalp, and extremities. Some patients show koilonychia. Spontaneous improvement or remission may occur at puberty or during pregnancy, suggesting a hormonal influence, but the condition may persist unchanged throughout adulthood. Administration of oral retinoids or 2% minoxidil have been reported to improve the alopecia.
Monilethrix most commonly is autosomal dominant and results from mutations in hair (or trichocyte) keratins. These hair keratins have a high cysteine content, making them “hard” keratins with a high degree of crosslinking. Of the 26 known hair follicle-specific keratins, three have been associated with monilethrix and all are type II keratins. Mutations in KRT81 and KRT86 are most common, but a mutation in KRT83 has been reported. Because keratins provide structural integrity to hair, abnormalities in these keratins lead to hair fragility with breakage occurring at the internodal sites. Mutations in hair keratins have also been linked to pure hair and nail ectodermal dysplasia (PHNED) ( KRT85 ), woolly hair with hypotrichosis ( KRT71 and KRT74 ), and pseudofolliculitis barbae ( KRT75 ). The majority of the hair keratins remain unlinked to human phenotypes but may impart variants in hair strength, texture, or curliness.
Autosomal recessive monilethrix has been linked to mutations in DSG4 , which encodes desmoglein 4, a transmembranous cell-adhesion molecule of the cadherin family that is predominantly expressed in the hair cortex and upper cuticle. Desmoglein 4 is thought to integrate keratin filaments into desmosomes. Desmoglein 4 mutations may also manifest as autosomal recessive hypotrichosis, which resembles monilethrix but lacks the characteristic beaded appearance of the hair shaft under light microscopy.
Pseudomonilethrix.
Pseudomonilethrix was originally described as an autosomal dominant developmental defect of fragile hair with irregularly shaped nodes. In fact, the hair changes are artifactual and related to overlapping hairs under the pressure of an overlying glass slide. Pseudomonilethrix is seen more commonly when fine hairs are handled by forceps.
Trichorrhexis Invaginata.
Trichorrhexis invaginata (bamboo hair) is characterized clinically by dry, lusterless, easily fractured, sparse, and short hair. Under light microscopy, the hairs show a peculiar intussusception or telescope-like invagination along the hair shaft, which microscopically resembles the ball-and-cup joints of bamboo. Variations in trichorrhexis invaginata occur, most commonly “golf-tee hair,” presenting the expanded proximal end of an invaginate node. The hair defect in trichorrhexis invaginata is thought to be abnormal keratinization of the hair shaft, which results in softening of the hair cortex and promotes intussusception of the distal portion of the hair shaft into the softer proximal portion.
Although trichorrhexis invaginata may occur as an isolated finding, this hair shaft abnormality is characteristic of Netherton syndrome ( Fig. 7-5 ), an autosomal recessive genodermatosis that has been linked to mutations in SPINK5 (see Chapter 5 ). Neonates with this disorder characteristically show generalized exfoliative erythroderma and failure to thrive, often associated with hypernatremic dehydration, recurrent infections, and sepsis. Severely affected neonates may show extremely sparse and even absent hair, making the diagnosis based on hair shaft examination difficult. However, the eyebrows are almost always short and broken (see Fig. 7-5, A ); eyebrow hairs should be examined in cases in which the abnormality cannot be demonstrated from scalp hair, and trichoscopy (dermoscopy) can be helpful.
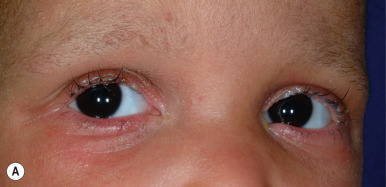
Beyond infancy, many affected individuals show the characteristic skin finding of ichthyosis linearis circumflexa, with walls of scale surrounding red patches, in addition to their dry, lusterless hair that breaks easily (see Fig. 7-5, B , and Figs. 5-20 and 5-21 ). Atopic conditions usually accompany the ichthyosis. Although spontaneous remission of the hair defect has been described (generally between 6 and 15 years of age), the vast majority show persistence.
Pili Torti.
Pili torti hairs show three or four regularly spaced twists that occur at irregular intervals along the hair shaft. The hair shaft appears flattened at the site of the twist, which is almost always through 180 degrees. The dry, fragile hair is often lighter in color than expected and shimmers in reflected light with a “spangled” appearance because of the hair twisting. The hair tends to be short, especially in areas subject to trauma, and may extend out from the scalp. Pili torti must be distinguished from twisted hair, which has been described in association with anorexia nervosa.
Pili torti may occur as an isolated phenomenon with onset at birth or early infancy. This genetic disorder shows both autosomal dominant and autosomal recessive inheritance patterns. The appearance of the hair in patients with pili torti may become more normal with time, although twisted hairs can still be found in the adult scalp; those who still manifest the disorder at puberty, however, are unlikely to show significant improvement with age. A late-onset autosomal dominant form has also been described, in which brittle hair and patchy alopecia develop after puberty. Mental retardation has been noted in some pedigrees. Pili torti has been associated with several mitochondrial disorders (see Pili Torti section).
Menkes syndrome (trichopoliodystrophy) is an X-linked recessive neurodegenerative disorder that affects male infants. Classic Menkes syndrome affects 90% to 95% of patients, with a less common mild form associated with long survival and occipital horn syndrome (previously called X-linked cutis laxa or Ehlers-Danlos syndrome type IX) showing largely connective tissue manifestations. Carrier females may exhibit pili torti. Classical Menkes syndrome is characterized by coarse facies; pili torti; temperature instability; seizures; psychomotor retardation; arterial intimal changes; soft, doughy skin; joint laxity; low or absent plasma copper and ceruloplasmin levels; growth failure; increased susceptibility to infection; and death, generally by age 3 or 4 years. Clinical features often include premature birth, hypothermia, and relatively normal development until 2 to 6 months of age, when drowsiness and lethargy are noted, intractable seizures begin, and growth and development cease. Rarely neonates demonstrate erythroderma as an early sign.
Usually the hair is fine, dull, sparse, and poorly pigmented in infancy; it stands on end and looks and feels like steel wool ( Fig. 7-6 ). Additional features include tortuosity of cerebral and other medium-sized arteries; osteoporosis; frequent subdural hematomas; widening of the metaphyses with spurring; and frequent fractures, at times simulating the radiologic findings characteristic of patients with battered child syndrome. Although pili torti is generally a prominent feature of this disorder, other less commonly reported hair abnormalities include monilethrix and trichorrhexis nodosa.
Menkes syndrome results from mutations in ATP7A, which encodes a copper-transporting adenosine triphosphatase (ATPase) that incorporates copper into copper-dependent enzymes and maintains copper levels by removing excessive copper from the cytosol. The combination of clinical features, bone abnormalities, and low plasma copper and ceruloplasmin levels establishes the correct diagnosis. Parenteral administration of copper histidine occasionally prevents neurologic degeneration and pigmentation if initiated in the neonatal or infantile period but often is ineffective.
Crandall syndrome, an X-linked recessive disorder, consists of pili torti with alopecia, sensorineural deafness, and hypopituitarism. Björnstad syndrome is characterized by sensorineural deafness, pili torti, and occasionally mental retardation. The syndrome is autosomal recessive, but families with an autosomal dominant pattern have been described. The recessive form has more recently been linked to mutations in BCS1L, which encodes an ATPase needed to assemble complex III in the mitochondria. More severe defects in BCS1L markedly increase reactive oxygen species and are lethal to neonatal infants with multisystemic involvement (complex II deficiency and growth retardation, aminoaciduria, cholestasis, iron overload, lactic acidosis, and early death [GRACILE] syndrome).
Bazex–Dupré–Christol and Rombo syndromes are X-linked dominant traits characterized by congenital hypotrichosis with pili torti, number facial milia, trichoepitheliomas, vellus hair cysts, and an increased risk of the early development of basal cell carcinomas. Distinguishing features are follicular atrophoderma, hypohidrosis, comedones and facial and neck pigmentation in Bazex–Dupré–Christol syndrome, and atrophoderma vermiculatum and photosensitivity in Rombo syndrome. Females often do not show the hypotrichosis, with normal and pili torti hairs intermingled.
Hypotrichosis with juvenile macular dystrophy (HJMD) is characterized by the development of sparse, short hair from birth or in the first months of life. Fusiform beading and pili torti may be seen by microscopy. Macular degeneration first develops in the first or second decade, leading to blindness by early adulthood. Patients suspected of having HJMD should have annual ophthalmologic evaluations, because early retinal pigmentation and atrophy precede the decrease in visual acuity. The disorder is autosomal recessive and caused by mutations in CDH3 , which encodes P-cadherin. CDH3 mutations also cause ectodermal dysplasia, ectrodactyly, and macular dystrophy ( Table 7-1 ).
| Disorder | Inheritance | Gene | Protein | Function |
|---|---|---|---|---|
| Group 1 | ||||
| TNF/TNFR Pathway | ||||
| Hypohidrotic ectodermal dysplasia | XLR | EDA1 | Ectodysplasin (EDA) | Membrane ligand |
| Hypohidrotic ectodermal dysplasia | AD | EDAR | EDA receptor (EDAR) | Receptor of EDA |
| EDARADD | EDAR-associated death domain | Adaptor molecule | ||
| Hypohidrotic ectodermal dysplasia | AR | EDAR | See above | See above |
| EDARADD | ||||
| TRAF6 | TNF receptor associated factor 6 | Activates IKK | ||
| NF-κB Inhibitors | ||||
| Hypohidrotic ectodermal dysplasia with immune deficiency (males) + osteopetrosis (males) | XLR | NEMO/IKKγ | NF-κB essential modulator | NF-κB activation |
| Incontinentia pigmenti (females) | XLD | NEMO/IKKγ | NF-κB essential modulator | NF-κB activation |
| Hypohidrotic ectodermal dysplasia with immune deficiency | AR | IκBα | IκBα | NF-κB activation |
| Transcription Factors | ||||
| Ectrodactyly-ectodermal dysplasia-clefting syndrome | AD | p63 | p63 | Transcription factor |
| Rapp–Hodgkin syndrome | AD | p63 | p63 | Transcription factor |
| Ankyloblepharon-ectodermal dysplasia-clefting syndrome (AEC) | AD | p63 | p63 | Transcription factor |
| Acrodermatoungual-lacrimal-tooth (ADULT) | AD | p63 | p63 | Transcription factor |
| Limb-mammary syndrome | AD | p63 | p63 | Transcription factor |
| Trichodentoosseous syndrome | AD | DLX3 | DLX3 | Transcription factor |
| Witkop syndrome | AD | MSX1 | MSX1 | Transcription factor |
| Ellis van Creveld syndrome | AR | EVC, EVC2 | EVC, EVC2 | Unknown |
| Group 2 | ||||
| Clouston syndrome | AD | GJB6 | Connexin 30 | Intercellular junctions |
| Oculodentodigital dysplasia (ODDD) | AD | GJA1 | Connexin 43 | Intercellular junctions |
| Clefting-ectodermal dysplasia | AR | PVRL1 | Nectin 1 | Interacts with cadherins, esp. at adherens junctions |
| Ectodermal dysplasia-syndactyly syndrome | AR | PVRL1 | Nectin 4 | Interacts with cadherins, especially at adherens junctions |
| Ectodermal dysplasia: skin fragility syndrome (see Chapter 13 ) | AR | PKP1 | Plakophilin 1 | Desmosomal plaque/stability |
| Ectodermal dysplasia, ectrodactyly, and macular dystrophy | AR | CDH3 | Cadherin 3/ P-cadherin | Adhesion molecule for cell–cell binding |
| Hypotrichosis with juvenile macular dystrophy | AR | CDH3 | Cadherin 3/ P-cadherin | Adhesion molecule for cell–cell binding |
| Odontoonychodermal dysplasia (OODD) | AR | WNT10A | Wnt10A | β-catenin-mediated signaling |
| Hypohidrotic ectodermal dysplasia | AR, AD | WNT10A | Wnt10A | β-catenin-mediated signaling |
| Schöpf–Schulz–Passarge syndrome | AR | WNT10A | Wnt10A | β-catenin-mediated signaling |
Individuals with mitochondrial enzyme abnormalities have shown a wide variety of abnormalities, predominantly failure to thrive and neuromuscular changes; however, skin or hair abnormalities have been described in 10% of affected children. Hair abnormalities range from alopecia to dry, thick brittle hair to hypertrichosis, especially on the back. Syndromic disorders with hair abnormalities may also affect mitochondrial function (e.g., Björnstad syndrome and cartilage-hair hypoplasia). Light-microscopy examination of affected hair has shown a variety of hair shaft defects associated with increased fragility including pili torti, TTD, trichorrhexis nodosa, and diffuse longitudinal grooving with flattened hair shafts. Patchy erythematous lesions have been described, and many of the patients with skin manifestations have shown mottled pigmentation.
Pili Bifurcati.
Pili bifurcati is an uncommon anomaly of hair growth characterized by intermittent bifurcation of the hair shaft in which affected hairs divide into two separate shafts that subsequently become rejoined along the hair shaft. This bifurcation is repeated at intervals, and the anomaly appears to be transitory, with only a small percentage of hairs exhibiting the bifurcation. This disorder should not be confused with pili multigemini, a disorder in which multiple hairs project from a single hair follicle.
Trichothiodystrophy.
TTD is a heterogeneous group of autosomal recessive disorders in which patients have dry, brittle, cysteine-deficient hair as an isolated finding or in association with often-multisystemic disease. To date, four genes have been linked to TTD: ERCC2 ( XPD ), ERCC3 ( XPB ), p8 or GTF2H5 ( TTDA ), and C7Orf11 ( TTDN1 ). The function of TTDN1 is not well understood, but it likely regulates cell cycling and transcription efficiency. The other three genes encode subunits of transcription/repair factor IIH (TFIIH), a multiprotein complex involved in transcription and nucleotide excision repair. Unlike xeroderma pigmentosum, TTD is not prone to cancer, although squamous cell carcinoma has been described.
Light microscopy of TTD hairs shows a wavy, irregular outline and a flattened shaft that twists like a folded ribbon. Two types of fracture may be seen: trichoschisis (clean transverse fracture) or an atypical trichorrhexis nodosa with only slight splaying of the cortical cells. Polarizing microscopy is critical to show the characteristic alternating light and dark bands, the “tiger-tail” appearance. Although the severity of hair shaft defects is inversely proportional to the hair sulfur content, there is no association between the extent of systemic disease and percentage of abnormal hairs. Sparse hair is often associated with the shaft defect. Some patients have described cyclic hair loss with fever, which may reflect a mutation leading to thermosensitive XPD .
A recent review of 112 published cases of TTD described the abnormalities beyond hair defects. Ichthyosis has been noted in 65% and clinical evidence of photosensitivity in 24% of these patients. The ichthyosis may resemble autosomal recessive congenital ichthyosis (ARCI) or ichthyosis vulgaris (see Chapter 5 ), and some patients show marked depletion of the granular layer in skin-biopsy sections. Of the patients who have TTD with ichthyosis, 37% show a collodion phenotype at birth. Patients may also show xerosis, palmoplantar keratoderma, atopic dermatitis, and/or follicular keratosis. Nail abnormalities have been described in 63% of patients overall, especially dystrophy with thickening or yellow discoloration.
Among the most common noncutaneous features are developmental delay/intellectual impairment (86%), short stature and low weight (73%), and ocular abnormalities (51%, especially cataracts). Facial dysmorphism is seen in 66% of patients, especially microcephaly, large or protruding ears, and micrognathia. Bone abnormalities are seen radiographically in 38%, particularly osteosclerosis and delayed bone age. Gonadal abnormalities were noted in 14% overall, most commonly hypogonadism and cryptorchidism. Recurrent infections have been noted in 46%, particularly involving the respiratory and gastrointestinal tracts and the inner ear, but were uncommonly associated with immunodeficiency or neutropenia. Overall, mortality in the first decade of life is increased 20-fold. Complications during pregnancy are noted in 26% of patients, most commonly intrauterine growth retardation, but also preterm delivery; preeclampsia; hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome; and prematurity.
Subgroups of TTD have been classified based on clinical characteristics (brittle hair, intellectual impairment, decreased fertility, and short stature [BIDS]; ichthyosis, brittle hair, infertility, developmental delay, short stature [IBIDS]/Tay syndrome, photosensitivity, ichthyosis, brittle hair, infertility, developmental delay, short stature [PIBIDS]). However, a new classification has been proposed that divides patients based on their mutations as group I (mutations in genes encoding subunits of TFIIH: XPD, XPB, p8 ), II ( TTDN1 ), and III (no known molecular basis). Group I includes patients with photosensitivity (either clinical or in vitro ) and is the most common subtype. Most individuals in group II are not photosensitive and show an increased risk of delayed bone age, seizures, and autistic behavior. Currently unclassified but nonphotosensitive patients (such as those with Pollitt and Sabinas syndromes) are in group III. Using this classification, ichthyosis and the collodion-baby phenotype are most highly correlated with group I, whereas hypogonadism has been found more in groups II and III.
Marie Unna Hypotrichosis.
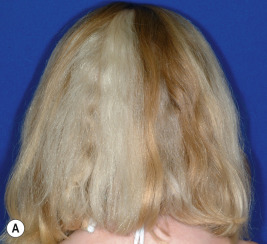
Marie Unna hypotrichosis is an autosomal dominant disorder manifested by almost complete congenital absence of scalp hair, eyebrows, and eyelashes. The hair regrows to normal density but is coarse, flattened, and twisted ( Fig. 7-7 ). Beginning at puberty the hair becomes progressively sparser, particularly on the vertex and scalp margins, resulting in a high frontal and nuchal hairline. By adulthood, only a sparse fringe of hair at the scalp margin may remain, and eyelashes, eyebrows, and body hair, including secondary-sexual hair, tend to be sparse. Scattered follicular horny plugs may be associated. Other ectodermal structures are unaffected, except that 50% of affected individuals show exceptionally widely spaced upper incisor teeth. Mutations that cause Marie Unna hypotrichosis affect U2HR, an open-reading frame upstream of the hairless gene that inhibits hairless expression; as a result, hairless expression and Wnt signaling are increased.

Hair Shaft Abnormalities Without Increased Fragility
Pili Annulati.
Pili annulati (ringed hair) is an autosomal dominant condition with onset shortly after birth. The hair looks shiny with attractive highlights, but alternating bright and dark bands are seen on close inspection. The bright areas are the result of light scattering from clusters of air-filled cavities within the cortex that appear as dark areas under light microscopy, especially in more proximal hair regions. Pili annulati may increase the risk of developing AA and has been reported to markedly improve or clear after the occurrence of alopecia totalis. The gene mutated in pili annulati has been linked to chromosome 12q24.33, but no mutations in candidate genes have been found.
Pseudopili annulati is an unusual variant of normal hair in which bright bands are seen at intervals along the hair shaft. Secondary to periodic twisting or curling of the hair shaft, this banding is conspicuous only in blond hairs and represents an attractive optical effect caused by reflection and refraction of light by flattened and twisted hair surfaces.
Woolly Hair
Woolly hair describes a tight, curly hair that is usually present from birth and shows abnormalities under light microscopy. The individual scalp hairs are fine and dry, light-colored, and corrugated at intervals, resembling the wool of sheep. Recognition of woolly hair is important because of the many associated abnormalities. The autosomal recessive disorders characterized by generalized woolly hair, keratoderma and dilated cardiomyopathy (Naxos disease and Carvajal syndrome) have been linked to mutations in desmoplakin and plakoglobin, two desmosomal proteins; keratoderma and woolly hair without cardiomyopathy may also result from mutations in KANK2 (see Chapter 5 ). Diffuse woolly hair has also been associated with ocular abnormalities, keratosis pilaris atrophicans and/or ulerythema ophryogenes ( Fig. 7-8 ), keratosis follicularis spinulosa decalvans (KFSD), giant axonal neuropathy (GAN) syndrome, and primary osteoma cutis.
Woolly hair without associated systemic manifestations can be inherited as either an autosomal dominant or autosomal recessive trait, both of which result from abnormalities of the inner-root sheath of the hair follicles. The entire scalp tends to be affected from birth, but nonscalp hair is normal. The hair grows slowly, is hypopigmented, and shows varying degrees of hypotrichosis. Plucked hair shows a dystrophic bulb and sometimes nonspecific shaft defects. The dominant form results from mutations in KRT71 or KRT74, encoding hair keratins 71 and 74, respectively.
The recessive forms have now been explained by mutations in two interacting genes, LPAR6 (encoding the lipophosphatidic acid [LPA] receptor 6/ P2RY5 ) and LIPH (encoding lipase H). Lipase H reduces phosphatidic acid to LPA, which is involved in lipid and energy metabolism. P2RY5 is thought to be a receptor for lysophosphatidic acid in the hair follicle.
The woolly hair nevus is a sporadic condition characterized by the development of one or more patches of hair different in color, shape, and consistency from the normal surrounding scalp hair ( Fig. 7-9 ). The hairs on the affected area are usually smaller in diameter, lighter in color, and sparser than those on the rest of the scalp. When examined under a dissecting microscope, the individual hairs are noted to twist about their long axis. The majority of reported cases of woolly hair nevus have been recognized during the first few months of life, but some have appeared in young adulthood. In about 50% of cases, woolly hair nevus coexists with a linear epidermal nevus in the same area or elsewhere. Ocular involvement has been described as an associated feature. Epidermal nevi with woolly hair have recently been linked to mutations in HRAS.
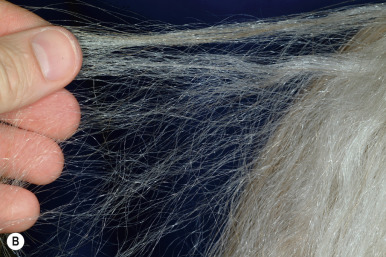
Acquired Progressive Kinking of the Hair.
Acquired progressive kinking of the hair is a rare disorder of scalp hair, with onset in adolescence or in young adulthood. The condition is characterized by a rapid onset of extreme curliness of the hair (mainly on the frontoparietal region of the scalp and vertex), often in association with an increased coarse texture, diminished luster, and striking unruliness. The disorder is more common in males than in females. Hair may become darker or remain unaltered in color, and the rate of growth may be decreased or unchanged. Examination of abnormal hairs by light microscopy reveals alterations in hair shaft diameter and partial twisting of the hair on its longitudinal axis.
Although the etiology of this disorder is unknown, it may follow treatment with systemic retinoids including isotretinoin. The localization of hair kinking, family history of androgenetic alopecia (AGA), pathologic features of affected scalp, and tendency to evolve into AGA suggest acquired kinking as a harbinger of AGA. No therapy is effective, and application of topical minoxidil has not affected the progressive thinning of hair in the areas of kinking. Spontaneous reversion to normal hair has been reported. Coarse hair but not woolly hair is also a feature of Hajdu–Cheney syndrome, resulting from mutations in NOTCH2. Other features include craniofacial abnormalities, short stature, acroosteolysis with broad nails and digits, synophrys, coarse skin, and premature tooth loss with periodontitis.
Uncombable Hair Syndrome.
The uncombable hair syndrome (pili trianguli canaliculi, spun-glass hair syndrome) is a unique hair disorder characterized by very pale, silvery, blond, or straw-colored hair. The hair is dry, frizzy, and unruly, and it does not lie flat on the scalp, thus making combing impossible ( Fig. 7-10 ). The syndrome is thought to be autosomal dominant with variable penetrance, although no associated gene mutations have been identified. The onset is usually during infancy or early childhood, and eyebrows, lashes, and body hair are normal. Affected children may have minor nail abnormalities and some show both uncombable hair and loose anagen hair (see Loose Anagen Syndrome section). The characteristic structural defect, the presence of canalicular depressions along the hair shaft, can be demonstrated by scanning electron microscopy or by routine microscopy of hair in cross-section. Cross-sectional microscopy shows a variety of shapes, including triangular, quadrangular, and reniform. The longitudinal grooving and abnormal shape in cross-section, however, are not specific for uncombable hair syndrome and have been described in several other syndromes, among them Marie Unna hypotrichosis, the ectodermal dysplasias with clefting, hypohidrotic ectodermal dysplasia (HED), oral-facial-digital syndrome type I (OFD1), and progeria. The clinical appearance of the spun-glass hair requires a sizable proportion of abnormal hairs, and at least 50% of hairs are abnormal by scanning electron microscopy. The hair tends to become progressively more manageable by adolescence, and some patients have responded to biotin administration.
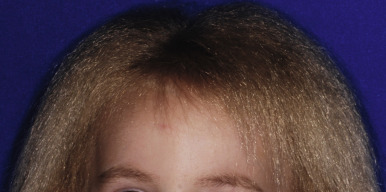
Uncombable hair syndrome must be distinguished from extremely unruly hair, which is seen in 2% of individuals. Extremely unruly hair that tends to stand up from the area of the posterior parietal whorl toward the frontal hairline may be associated with microcephaly and is a potential indicator of abnormal brain growth and morphogenesis, similar to upsweep of anterior scalp hair and aberrant parietal whorl position.
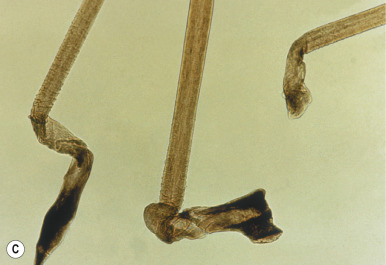
Loose Anagen Syndrome
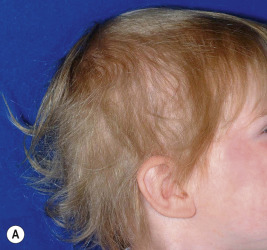
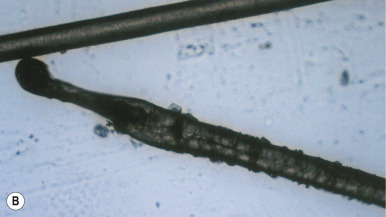
Loose anagen syndrome occurs in 10% of all children who have alopecia and is characterized by actively growing anagen hairs that are loosely anchored and can be easily and painlessly pulled from the scalp. Although considered an autosomal dominant disorder, most cases are sporadic and occur in girls. A mutation in a hair keratin ( KRT75 , formerly called K6hf ) has been found in some families, but its relevance is unclear because it does not reliably segregate with the loose anagen phenotype. Most patients are blond girls above the age of 2 years (mean, 6 years of age). Affected children generally have sparse, short, scalp hairs that seldom require cutting ( Fig. 7-11, A ). Examination shows patchy or subtle diffuse thinning with hairs of uneven length. The hair often appears to be limp, and a matted texture has been noted, particularly in occipital hair. Of the actively growing anagen hairs, more than 80% show ruffled cuticles and pigmented misshapen bulbs ( Figs. 7-11, B and C ). Gentle pulling tends to yield several hairs, allowing the diagnosis to be made by light-microscopic examination of hair; forceful extraction of hairs may lead to misshaping of normal anagen hairs and thus should be avoided. Shedding of the hair is cyclic, and the inability to extract large amounts of hair by gentle pull test does not definitively rule out the diagnosis. Although no treatment is available for this disorder, it is reassuring for patients and their families to know that other abnormalities are not associated with this disorder and individuals with this condition tend to improve with time.
Loose anagen hair is also a feature of an autosomal recessive Noonan-like syndrome (Noonan-like syndrome with loose anagen hair) that has been linked to mutations in SHOC2. In addition to the fine, sparse hair, affected children show the Noonan syndrome facies and broad neck; macrocephaly; reduced growth with delayed bone age (often from growth hormone deficiency); variable cognitive defects, hyperactivity; hypernasal voice; darkly pigmented, thickened skin with dermatitis; and cardiac defects.
Nonscarring Alopecias Without Hair Shaft Abnormalities
Congenital/Genetic Disorders
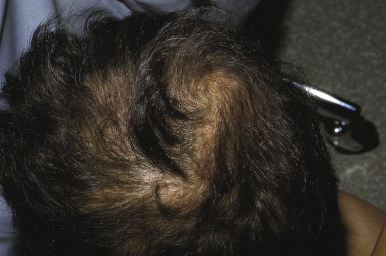
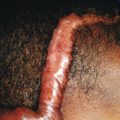
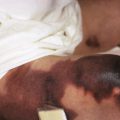
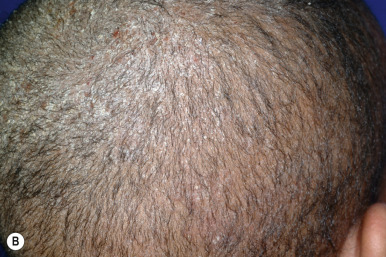
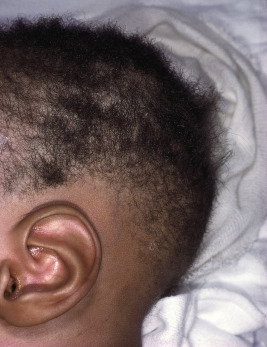
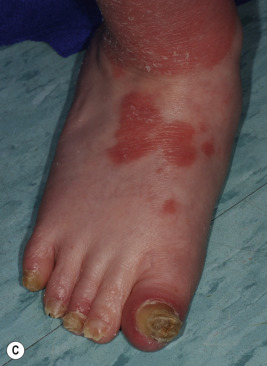
Congenital triangular alopecia is characterized by an area of alopecia that, although sometimes notable at birth in babies with abundant scalp hair, is usually detected at 2 or 3 years of age ( Fig. 7-12 ). Initial appearance during adulthood has been described. The area is triangular and overlies the frontotemporal suture, with the base of the triangle directed forward. The triangular patch may extend to the hairline, but often a fringe of hair may separate it from the forehead. Generally measuring 3 to 5 cm from base to apex, the area may be completely bald or partially covered by vellus hairs and remains unchanged throughout life. Dermoscopy of the affected area shows normal follicular openings with vellus hairs, whereas dermoscopy of AA shows dystrophic and exclamation-point hairs. Although unilateral in 80% of affected individuals, it may be bilaterally symmetric, and on rare occasions similar triangular patches may be noted on the nape of the neck.
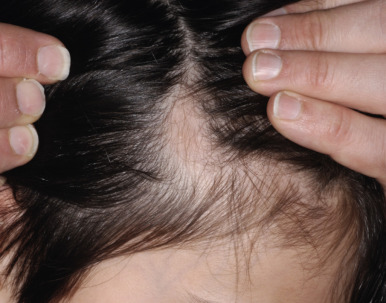
The condition is almost always sporadic, although the association with developmental delay and seizures has been described in a mother and daughter. Congenital triangular alopecia has been described in a patient each with phakomatosis pigmentovascularis, Down syndrome, and Dandy–Walker malformation. Hair transplants have been used to repopulate the area of triangular alopecia. Bilateral congenital localized patches of alopecia of the parietal area that resemble the alopecia of congenital triangular alopecia have been seen in patients with Gomez–Lopez–Hernandez syndrome (cerebellotrigeminal–dermal dysplasia). Although the alopecia classically affects the parietal area, other sites have been reported to show symmetric alopecia. Other features are skull defects (craniosynostosis with brachycephaly, midfacial hypoplasia), neurologic abnormalities, short stature, hypertelorism, and corneal opacities.
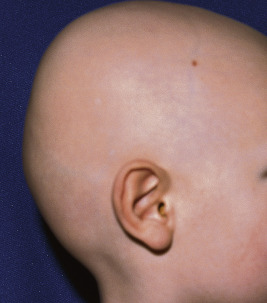
Individuals who initially have hair but lose it all with the first hair shedding shortly after birth likely have atrichia with papular lesions (APL; resulting from mutations in the hairless gene) or vitamin D-dependent rickets type IIA (caused by mutations in the vitamin D receptor). Affected individuals never regrow scalp hair and tend to be nearly totally devoid of eyebrows, lashes, and axillary and pubic hair ( Fig. 7-13 ). Follicular cysts and milia-like lesions may appear on the skin later in life (hence the nomenclature of with papular lesions ). Scalp biopsies show disintegration of the lower two-thirds of the hair follicle, which is often replaced by cysts. Patients with vitamin D-resistant rickets are clinically and histologically identical to patients with APL but show the additional manifestations of early-onset rickets, hypocalcemia, secondary hyperparathyroidism, and elevated 1,25-dihydroxyvitamin D 3 . The similarity in phenotype reflects the direct regulatory effect of the vitamin D receptor and hairless gene on each other via a transcriptional mechanism. This group of patients is often misdiagnosed as having alopecia universalis, and they must be also be distinguished from patients with PHNED, an autosomal recessive disorder resulting from mutations in KRT85, KRT74, or the HOXC13 homeobox gene, which usually presents with congenital complete alopecia and uniformly small dystrophic nails. Sometimes, very short, sparse, fragile hair may be present in PHNED and is easily visualized by trichoscopy.
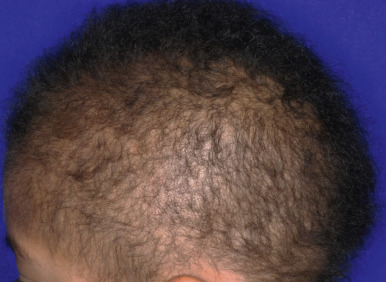
Hypotrichosis Simplex
Hypotrichosis simplex is a group of rare autosomal dominant and autosomal recessive nonscarring alopecias in which patients are usually born with normal hair. Hair loss can begin in the first months or even as late as the first decade ( Fig. 7-14 ) and can progress to almost complete loss of scalp hair by adulthood. Graying has been reported to coincide with hair loss. Some individuals show sparse, fine, short hairs, especially at the crown, but hair on sites other than the scalp is normal. The disorder has considerable genetic heterogeneity. Autosomal dominant forms result from mutations in genes encoding corneodesmin, a protein of the corneocyte desmosomes and inner hair sheath (see Peeling Skin Syndrome section, Chapter 5 ) ; a protein of the pre-messenger-ribonucleic acid (mRNA) processing spliceosome ( SNRPE ) ; and APCDD1, an inhibitor of Wnt pathway signaling. Autosomal recessive forms of hypotrichosis simplex can include hypotrichosis simplex with woolly hair (see Woolly Hair section) or be associated with scalp follicular keratosis, which can be confused with monilethrix.
Hallermann–Streiff, Sensenbrenner, Coffin–Siris, and growth retardation, alopecia, pseudoanodontia, and optic atrophy (GAPO) syndromes all show hypotrichosis in association with facial dysmorphism and other physical signs (see Online only).
In the Hallermann–Streiff syndrome (oculomandibulodyscephaly), hypotrichosis of the scalp, eyebrows, and eyelids is associated with dwarfism, beaked nose, and brachycephaly. The alopecia is most prominent at the frontal and parietal areas and is especially marked along suture lines. Axillary and pubic hair may also be scant, and cutaneous atrophy, largely limited to the scalp and nose, may appear as thin, taut skin and prominent underlying blood vessels. Other features include frontal and parietal bossing, mandibular hypoplasia, microphthalmia, low-set ears, thin and small lips, high-arched palate, atrophy of the skin of the face, congenital cataracts, blue sclerae, motor and occasionally mental retardation, and dental abnormalities.
Sensenbrenner syndrome, or cranioectodermal dysplasia, is a rare autosomal recessive disorder manifested by small stature, dolichocephaly, an unusual facies, and tubulointerstitial nephritis leading to early end-stage renal failure. The typical facies show frontal bossing, hypertelorism, prominent epicanthal folds, antimongoloid palpebral fissures, eversion of the lip, and full-rounded cheeks. Patients have small, gray, widely spaced teeth; short, fine, hair; and hypohidrosis. Sensenbrenner syndrome is a ciliopathy that results from mutations in IFT122, WDR35, or IFT43.
Patients with the Coffin–Siris syndrome most commonly show a constellation of severe mental retardation; a characteristic coarse-appearing facies; scalp hypotrichosis with hypertrichosis of the eyebrows, eyelashes, face, and back; hypotonia; hypoplastic to absent fifth fingernails and distal phalanges; and feeding problems with postnatal growth deficiency. Coffin–Siris syndrome is autosomal dominant and results from mutations in one of several components of the SWItch/sucrose nonfermenting (SWI/SNF; also called BAF ) adenosine triphosphate (ATP)-dependent chromatin-remodeling complex. Of these, mutations in ARID1B are most common (76% in one series).
GAPO is an acronym for growth retardation, alopecia, pseudoanodontia, and optic atrophy . Distinctive craniofacial features include frontal bossing, high forehead, mid-facial hypoplasia, hypertelorism, and thickened eyelids and lips. GAPO syndrome is autosomal recessive and results from mutations in ANTXR1, which encodes anthrax toxin receptor 1.
Cartilage-hair hypoplasia syndrome is an autosomal recessive disorder that occurs primarily in inbred Amish or Finnish populations. Patients have short limbs and sparse, fine scalp and body hair. Several patients with hyperextensible digits and soft, doughy skin, reflecting degenerated elastic tissue, have been described. Defective cell-mediated immunity is seen in most patients and results in relative anergy, altered T-cell responses, and increased susceptibility to severe viral infections, particularly varicella. Patients may have infantile neutropenia, Diamond–Blackfan anemia, severe combined immunodeficiency, celiac syndrome, and/or toxic megacolon. Mild to severe bronchiectasis has been noted in more than 50% of patients. Approximately 10% develop malignancy, especially lymphoreticular; an increased prevalence of early basal cell carcinomas has also been described. The disorder results from mutations in ribonuclease (RNase) MRP, which cleaves RNA in mitochondrial deoxyribonucleic acid (DNA) synthesis and preribosomal RNA in the nucleolus. Alopecia is seen with macrocephaly, cutis laxa, and scoliosis in macrocephaly, alopecia, cutis laxa, and scoliosis (MACS) syndrome (see Chapter 6 ).
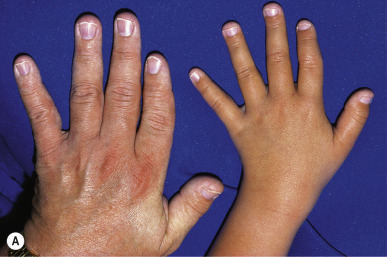
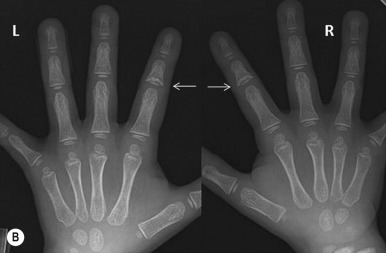
Trichorhinophalangeal syndrome (TRPS) type I is an autosomal dominant disorder characterized by a distinctive facies with pear-shaped nose, elongated philtrum, thin upper lip, supernumerary incisors, and receding chin ( Fig. 7-15 ) and skeletal abnormalities including brachydactyly, deviation of the middle phalanges ( Fig. 7-16, A ), hip malformation, and short stature. Most patients show fine, sparse, slow-growing hair, but almost-normal hair to complete baldness have been described. The underlying molecular basis is mutation in TRPS1, which encodes a transcription factor. Individuals with type II TRPS (Langer–Giedion syndrome) have associated multiple cartilaginous exostoses. The diagnosis is made by the demonstration of cone-shaped epiphyses of the fingers seen on plain radiography ( Fig. 7-16, B ). These findings may not be detectable until 3 years of age or older. The type II form is a contiguous gene syndrome, with deletion of both the TRPS1 gene and the gene that is mutated in multiple exostosis type I ( EXT1 ). Type III TRPS results from mutations in TRPS1 but manifests with much more severe short stature and generalized shortening of all phalanges and metacarpals than TRPS type I.
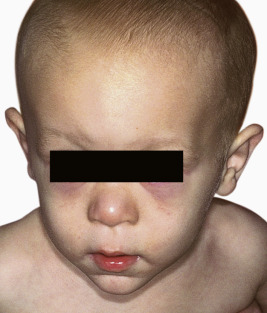
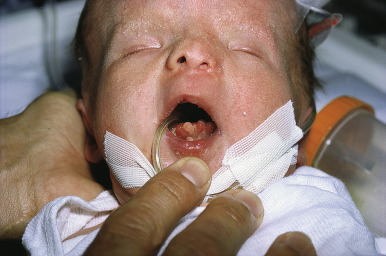
OFD1 is an X-linked dominant disorder limited to girls and thought to be lethal in boys. Facial features occur in almost 70% of patients and include hypoplasia of nasal cartilages and hypertelorism with lateral displacement of the inner canthi (dystopia canthorum) ( Fig. 7-17 ). Among the oral anomalies described are tongue hamartomas, lobulated cleft tongue, cleft lip and palate, maldeveloped frenula, asymmetry of the lips and tongue, and maxillary gingival swelling. Associated hand malformations are common and include brachydactyly, syndactyly, clinodactyly, and polydactyly. Almost half of affected individuals show central nervous system (CNS) involvement, most commonly retardation or selective cognitive impairment. Cutaneous abnormalities occur in the minority of patients but include numerous milia at birth and sparse fine or coarse, dry and lusterless hair to frank alopecia. Polycystic kidney disease with renal insufficiency is occasionally seen in children but more often occurs with advancing age (>50% after age 36 years). The disorder results from mutations in OFD1, which encodes a centrosomal protein of primary cilia. Other “ciliopathies” share the CNS, skeletal, and cystic renal abnormalities of OFD1.
Ectodermal Dysplasias
Ectodermal dysplasias are a complex group of approximately 200 developmental disorders that were traditionally classified based on their sites of abnormalities (hair, teeth, nails, and/or eccrine glands) and other ectodermal and nonectodermal features. In 2009 Priolo suggested a new classification of ectodermal dysplasias that focuses on the molecular basis of these disorders (now known in approximately 80 of the ectodermal dysplasias), the function of the affected proteins, and the clinical features. As a result, ectodermal dysplasias are divided into two groups (see Table 7-1 ). The first group includes disorders in which ectodermal derivatives fail to develop or differentiate because of the absence of reciprocal signals from the ectoderm to the mesenchyme. This group can be further divided into: (1) abnormalities of the tumor necrosis factor (TNF)-like/TNF receptor pathway (including HED); (2) the nuclear factor κB (NF-κB), NF-κB essential modulator (NEMO), and inhibitor of κB (IκB) molecules (such as HED with immunodeficiency [HED-ID]); and (3) transcription factors such as p63 and DLX3. Inductive signals for normal differentiation are preserved in the second group, but tissues become dysplastic because abnormal regulation of transcription or expression leads to altered cell–cell interactions or disorganization of the cytoskeleton. Abnormalities in group 2 may affect nectins (e.g., cleft lip/palate–ectodermal dysplasia syndrome), connexins (as in Clouston syndrome and oculodentodigital dysplasia), desmosomal proteins (such as plakophilin in ectodermal dysplasia/skin fragility syndrome, also classified as a form of epidermolysis bullosa simplex, see Chapter 13 ), and molecules that interact with β-catenins (cadherins and WNT10A, as in odontoonychodermal dysplasia [OODD]).
Group 1 Ectodermal Dysplasia
Hypohidrotic Ectodermal Dysplasia.
HED is characterized by the triad of reduced sweating, hypotrichosis, and defective dentition. The majority of affected individuals are male, and overall 92% have a mutations in one of four genes (see Table 7-1 ): ectodysplasin-A1 ( EDA1 at Xq12-q13) in 58%; its ectodysplasin receptor ( EDAR at 2q13) in 16%; Wnt10A ( WNT10A at 2q35) in 16% (classified with group 2); and EDAR-associated death domain ( EDARADD at 1q42.2-q43) in 2%. HED may also result from a heterozygous mutation in TRAF6, which is upstream of NEMO and promotes NF-κB activity, and in X-chromosome located XEDAR, encoding the receptor for EDA2, a different EDA isoform that binds to Traf6. Female carriers with an EDA1 mutation show random inactivation of the abnormal gene and can show manifestations ranging from none to extensive dental defects, alopecia, and patchy hypohidrosis following the lines of Blaschko, which are lines of embryologic development of skin. Mutations in the receptor ( EDAR ), the death domain ( EDARADD ), or WNT10A ( WNT10A ) may be inherited in a recessive or dominant pattern. The phenotype with recessive mutations closely resembles those in X-linked recessive HED, whereas dominant mutations tend to be less severe with respect to sweating and hair loss. Mutations in WNT10A can lead to a spectrum of ectodermal defects that can include isolated oligodontia, mild manifestations of HED, OODD, or Schöpf–Schulz–Passarge syndrome. The nail changes of OODD (fragility, longitudinal ridging, splitting, koilonychias, onycholysis, and pterygium) begin in early childhood are associated with palmoplantar keratoderma, agenesis of the permanent teeth, and sometimes facial erythema and atrophic tongue papillae. Schöpf–Schulz–Passarge syndrome may manifest solely with bilateral eyelid cysts and palmoplantar keratoderma but often includes nail, hair, and/or dental abnormalities.
HED-ID affects 1:250,000 births and is usually caused by hypomorphic mutations in NEMO, an X-linked gene. Autosomal dominant HED-ID has been reported in six patients and results from hypermorphic (gain-of-function) mutations in IKBA . NEMO mutations cause incontinentia pigmenti in girls (see Chapter 11 ) and amorphic (i.e., there is effectively no gene product) tend to be lethal in males. Features of HED have been described in 77% of boys with immunodeficiency and NEMO mutations; osteopetrosis and lymphedema have been noted in 8% with HED-ID. Boys with HED-ID occasionally show the clinical vesiculopapules and histologic features of incontinentia pigmenti, but the distribution is not blaschkoid given the lack of mosaicism. Most mutations in NEMO that lead to HED-ID occur in exon 10 and affect the C-terminal zinc finger domain, which is critical for normal dendritic cell immune stimulation, but mutations leading to HED-ID are scattered throughout the NEMO gene.
Features of Ectodermal Dysplasia.
Affected persons often appear more like each other than like their own unaffected siblings ; classic features are usually obvious by infancy. Most have a distinctive pathognomonic facies: a square forehead with frontal bossing, large conspicuous nostrils, wide cheekbones with flat malar ridges, a thick everted lower lip, and a prominent chin. Ears may be small, satyr-like (pointed), low-lying, and anteriorly placed ( Figs. 7-18, A , and 7-19 ). Alopecia is often the first feature to attract attention but is seldom complete; overall 80% of males with the X-linked type have sparse hair. The hair also tends to be lightly pigmented and short. The skin is soft, thin, and light-colored but shows fine wrinkling and sometimes darkening of the periorbital areas. Many affected neonates are born with red, peeling skin, but collodion-like thickening has occasionally been described. Atopic dermatitis and other atopic conditions occur with increased incidence, and periorbital dermatitis is particularly common. The paucity of nail changes helps to distinguish HED from other ectodermal dysplasias, although nail changes were described in up to 50% of males and female carriers in a recent self-reported survey.
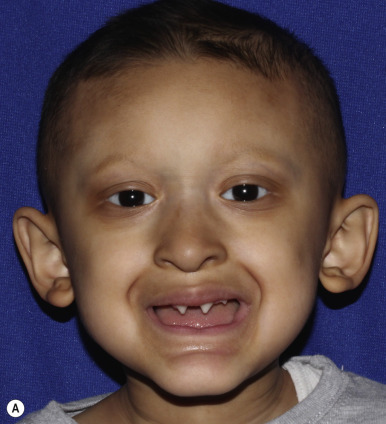
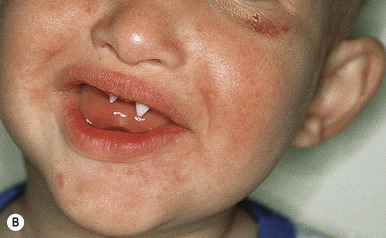
The decreased capacity for perspiration occurs in virtually all affected males and has a profound effect on life quality. Hypohidrosis often results in hyperthermia, and patients manifest with intermittent fevers, especially during hot weather or after exercise or meals. These recurrent fevers of unknown origin may be the presenting manifestation in affected infants. Hypoplastic lacrimal and mucous glands can lead to decreased tearing or epiphora, chronic nasal discharge, and an increased risk of otitis media and respiratory tract infections. Females and female carriers may have breast hypoplasia. Dentition is generally delayed, and dental anomalies vary from complete to partial absence of teeth with peg-shaped or conical incisors (see Fig. 7-18, B ).
Features of Immunodeficiency.
Boys with HED-ID usually have recurrent infections. Serious pyogenic infections occur in 86% of affected individuals, and mycobacterial infections (especially atypical Mycobacterium avium ) in 44%. Pneumocystis and viral and candidal infections occur less often. Bacteremia or sepsis is common, and the most common sites of infection are the lungs, sometimes leading to bronchiectasis, and the skin, sometimes with abscesses. Inflammatory colitis affects 21% of boys and presents as intractable diarrhea and/or failure to thrive. Some patients develop autoimmune hemolytic anemia. Natural killer-cell dysfunction has been described in all patients, but otherwise a range of immune defects have been described, largely reflecting the functional impairment in CD40, interleukin (IL)-1, TNF-α, and toll receptor signaling. Almost 60% of affected boys show hypogammaglobulinemia, with high levels of immunoglobulin (Ig) M in 15%. Hyper-IgM syndrome much more commonly results from mutations in CD40 ligand ( CD40L ) and may manifest with CD40L deficiency as oral aphthae and warts. Individuals with HED-ID are at high risk for early death from infections without transplantation.
Therapy for patients with all forms of HED is directed toward temperature regulation; cool baths and drenching with water, air conditioning, light clothing, cooling suits, and the reduction of the causes of normal perspiration are beneficial. Lubricating eye drops and nasal irrigation can compensate for the decreased glandular secretion. Minoxidil may promote some hair growth with long-term use. Dental intervention should begin by 2 years of age and can include dental prostheses and dental implants in older adolescents and adults to improve mastication, encourage normal speech development, and reduce cosmetic disfigurement. In mouse and dog models of X-linked HED, the prenatal or perinatal administration of recombinant Fc-EDA protein or ligand replacement using an EDAR -agonist antibody can improve dentition, lacrimation, sweating, and bronchopulmonary gland function, whereas postnatal administration in affected adult mice only improves sebaceous gland activity. Most recently, a single injection of EDA1 replacement protein into the amniotic fluid of pregnant eda1 -mutant mice provided a depot for recurrent oral ingestion of affected offspring without maternal absorption; treatment led to darker, denser coat; normal eye opening, tail shape, and teeth; and functional sweat glands. Prenatal diagnosis has been made noninvasively by tooth-germ ultrasound. The National Foundation for Ectodermal Dysplasias (NFED) provides excellent education materials on management of the hypohidrosis and dental abnormalities.
P63 -Related Forms of Ectodermal Dysplasia.
Mutations in p63, a gene that plays a critical role in maturation of ectodermal, orofacial, and limb development, lead to an autosomal dominant disorder with ectodermal dysplasia, orofacial clefting, and limb malformations as key characteristics. These clinical manifestations have traditionally been used to classify subtypes, but significant clinical and genotypic overlap is now recognized. Included are Rapp–Hodgkin syndrome (clefts of the lip, palate, and/or uvula; small narrow dysplastic nails; hypodontia with small conical teeth; and maxillary hypoplasia) ; ankyloblepharon-ectodermal dysplasia-clefting (AEC or Hay–Wells syndrome; ankyloblepharon or congenital fusion of the eyelids in association with facial clefting and midfacial hypoplasia) ; ectrodactyly, ectodermal dysplasia, and cleft lip/palate (EEC) syndrome; limb-mammary syndrome (ectrodactyly, cleft palate, and mammary gland abnormalities); acrodermatoungual-lacrimal-tooth (ADULT) syndrome syndrome; and nonsyndromic split hand/foot malformation.
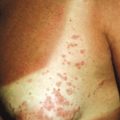
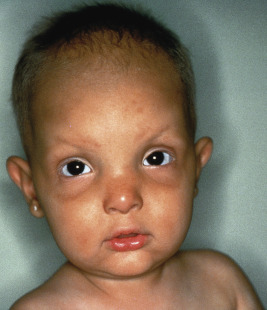
The skin, hair, teeth, nails, and glands (eccrine, sebaceous, lacrimal, mammary) are abnormally developed. The skin tends to be dry, itchy and hypopigmented. Extensive erosions have been described in 80% of neonates with the AEC phenotype. The hair is often sparse and wiry, and nails tend to be dystrophic. Scalp dermatitis and erosions with secondary chronic staphylococcal infection may be recurrent in the first few years of life and lead to cicatricial alopecia, especially of the vertex and frontal scalp ( Fig. 7-20 ). Teeth are often decreased in number with malformations and enamel hypoplasia. Hypohidrosis may be present, tearing is often decreased, and nipple hypoplasia has been described. Patients may show split hand/foot malformations (lobster claw deformity; ectrodactyly) and/or syndactyly. Short stature, poor weight gain, and hypospadias are other commonly described characteristics. Ectodermal dysplasia with clefting and/or ectrodactyly/syndactyly can be caused by mutations in a variety of other genes, i.e., other disorders (see Table 7-1 ). Disorders with p63 mutations also should be distinguished from CHAND syndrome or CHANDS, characterized by curly hair from birth, ankyloblepharon, nail dysplasia and, variably, ataxia ; the underlying genetic basis is unclear.
The trichodentoosseous syndrome is an autosomal dominant disorder characterized by kinky, curly hair at birth that tends to become straighter during childhood; small, widely spaced, pitted, eroded, and discolored teeth with early caries as a result of defective enamel; thickness and splitting of the nails; dolichocephaly, frontal bossing, and a square jaw, giving affected persons a distinctive facies; normal physical development; and increased bone density, especially of the cranial bones. The condition results from mutations in DLX3 , a crucial regulator of hair follicle differentiation and cycling.
The trichodental syndrome, also known as Witkop syndrome or the tooth-and-nail syndrome, is an autosomal dominant disorder characterized by fine, dry, slow-growing lusterless hair; sparseness or absence of the lateral halves of the eyebrows; congenitally missing and small teeth; and slow-growing, small, spoon-shaped nails (especially toenails) ( Fig. 7-21 ). Mutations have been described in MSX1, which directs the formation of teeth and nails. The other ectodermal dysplasia with mucocutaneous features included in group 1 is Ellis van Creveld syndrome. This autosomal recessive disorder features nail dysplasia in association with chondrodysplasia, polydactyly, orofacial abnormalities, and sometimes cardiovascular malformations. The mutated genes, EVC and EVC2, localize to cilia and are thought to be involved in hedgehog signaling.
Group 2 Ectodermal Dysplasia
Clouston Syndrome (Hidrotic Ectodermal Dysplasia).
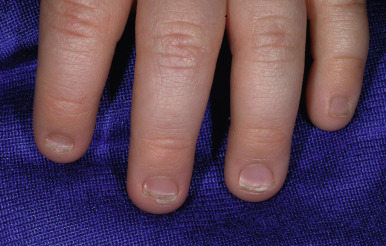
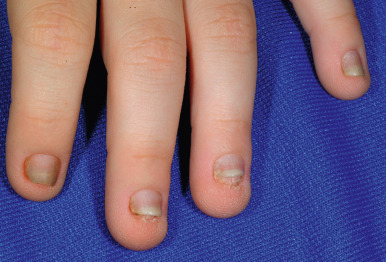
The most common hidrotic type of ectodermal dysplasia is Clouston syndrome, an autosomal dominant disorder characterized by nail dystrophy, hyperkeratosis of the palms and soles, and hair defects. Most cases have been reported in French-Canadian families. Unlike HED, individuals with the hidrotic form have a normal facies and show no abnormality of sweating, although eccrine syringofibroadenomas have been described. The teeth develop normally but are prone to caries. The predominant feature is congenital nail dystrophy, which may be the only manifestation in about one-third of affected individuals. The nails are thickened or thinned, striated, and often discolored ( Fig. 7-22 ). They may resemble the nails of pachyonychia congenita (PC) and are difficult to distinguish without genotyping until other features develop such as the painful character of the plantar keratoderma of PC, which is often present by 5 years of age, and the alopecia of hidrotic ectodermal dysplasia. The nails grow slowly and commonly show chronic paronychial infections that may result in partial to complete destruction of the nail matrix. Typically the skin and soft tissue surrounding the nail and at the finger pad appear thickened and swollen, leading to the term drumstick fingers. The palmoplantar keratoderma can extend to the dorsal aspects of the hands and feet. Hair may be normal during infancy and childhood but thereafter often becomes sparse, fine, and brittle and may eventuate in total alopecia ( Fig. 7-23 ). Body hair may be sparse; eyebrows and eyelashes may be thinned or absent. The skin may show a mottled hyperpigmentation with thickening and hyperpigmentation over the knees, elbows, and knuckles. Ocular abnormalities may include strabismus, conjunctivitis, and premature cataracts. Clouston syndrome has been attributed to mutations in GJB6, encoding connexin 30, a structural component of the intercellular gap junction. One patient with Clouston syndrome and bigenic mutations (one of each allele) of GJB6 and GJA1 (encoding connexin 43) have also been described. Mutations in GJB6 have also been noted in patients with keratitis, congenital ichthyosis, and neurosensory deafness (KID) syndrome (see Chapter 5 ), who share the palmoplantar keratoderma and sometimes early alopecia with thickening of the scalp. Clouston syndrome should also be distinguished from an autosomal recessive disorder that results from mutations in encoding grainyhead-like 2 ( GRHL2 ); in addition to nail dystrophy or loss and marginal palmoplantar keratoderma, patients show hypodontia and enamel hypoplasia, oral hyperpigmentation, dysphagia, and sometimes deafness or asthma. The combination of tretinoin and minoxidil has reportedly caused hair growth in Clouston syndrome.
Oculodentodigital Dysplasia.
Oculodentodigital dysplasia (ODDD), an autosomal dominant disorder, results from mutations in GJA1, which encodes connexin 43 ; mutations in GJA1 have also been linked to erythrokeratodermia variabilis (see Chapter 5 ). In addition to abnormalities of the eyes, teeth, and digits, patients show curly hair (sometimes with trichorrhexis nodosa), focal keratoderma, a characteristic facies with hypoplastic ala nasi, and neurologic, cardiac, and hearing defects. Cleft lip/palate-ectodermal dysplasia features spoon-shaped, slow-growing fingernails and toenails, pili torti, mental retardation, malformed ears, and partial syndactyly. It results from mutations in nectin 1, encoded by PVRL1 . Mutations in nectin 4, encoded by PVRL4, lead to ectodermal dysplasia-syndactyly syndrome (EDSS), characterized by alopecia, tooth abnormalities, and syndactyly.
Disorders of Follicular Plugging
Keratosis Pilaris
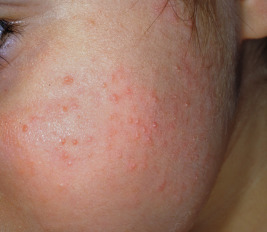
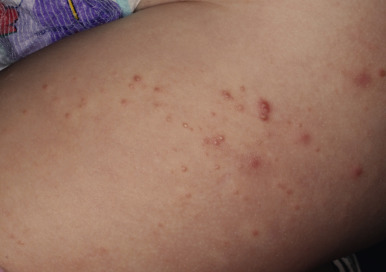
Keratosis pilaris is a common skin condition characterized by keratinous plugs in the follicular orifices surrounded by a variable degree of erythema (see Chapter 3 ). These small follicular-based papules are most commonly distributed on the cheeks ( Fig. 7-24 ), extensor areas of the upper arms, and anterior and lateral thighs ( Fig. 7-25 ) but may be widespread. Children with keratosis pilaris tend to have xerosis and sometimes atopic dermatitis and/or ichthyosis vulgaris. Occasionally, facial keratosis pilaris overlies intense erythema (keratosis pilaris rubra) and may also be pigmented (erythromelanosis follicularis faciei et colli). Keratosis pilaris does not tend to be symptomatic but may be cosmetically distressing, especially if quite inflammatory or extensive. Treatment is difficult but usually requires application of keratolytic agents such as creams or lotions containing lactic acid, glycolic acid, salicylic acid, or urea, and gentle exfoliation by a pumice stone, washcloth, loofah sponge, or Buf-Puf. Responsive patients must maintain therapy to achieve continued remission or improvement. The intense erythema of keratosis pilaris rubra may be lessened by pulsed-dye laser therapy or photopneumatic (vacuum-assisted pulsed-light) therapy. The erythema is sometimes decreased by treatment with low-strength topical steroids or calcineurin inhibitors.
Keratosis Pilaris Atrophicans
Numerous terms have been used to describe a group of interrelated syndromes characterized by inflammatory keratotic follicular papules and later by atrophy. Commonly described as atrophic variants of keratosis pilaris, these include ulerythema ophryogenes, atrophoderma vermiculata, and KFSD (keratosis pilaris decalvans) ( Table 7-2 ). This group of disorders has been attributed to abnormal keratinization of the follicular infundibulum, resulting in obstruction of the growing hair shaft, chronic inflammation, and scarring. No therapy is terribly effective, although topical keratolytic and anti-inflammatory agents (topical corticosteroids and calcineurin inhibitors) may reduce the keratotic and inflammatory components, respectively. In general, systemic retinoids have not been helpful. Durable eyebrow reconstruction using individual hair follicle micrografts in an adult with quiescent disease has been reported.
| Atrophoderma Vermiculata | Ulerythema Ophryogenes | Keratosis Follicularis Spinulosa Decalvans | |
|---|---|---|---|
| Skin lesions | Erythematous papules, follicular plugs, horn cysts, atrophic | Follicular papules, plugging, scarring | Milia, thorn-like follicular projections, atrophic scars |
| Sites | Cheeks, neck, limbs | Lateral eyebrows, extending medially | Scalp, eyebrows, eyelashes, cheeks, nose, neck, dorsal hands, fingers |
| Alopecia | Absent | Minimal eyebrows | Scarring alopecia of the scalp |
| Photophobia | Absent | Absent | Marked, corneal opacities |
| Inheritance | Sporadic or autosomal dominant | Sporadic or autosomal dominant | X-linked recessive or autosomal dominant |
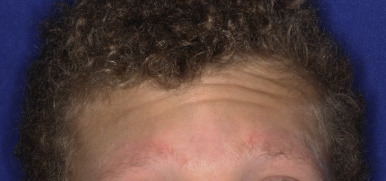
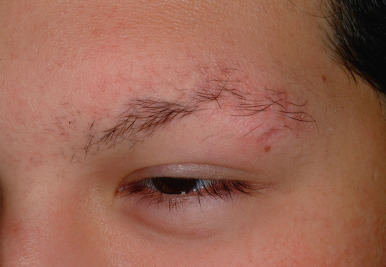
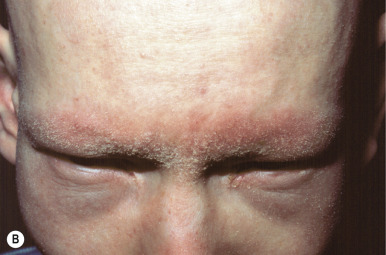
Ulerythema ophryogenes (keratosis pilaris atrophicans faciei) is characterized by persistent reticular erythema, small horny papules, atrophy, and scarring of the outer half of the eyebrows (see Fig. 7-8 ; Fig. 7-26 ). The disorder is more common in boys and usually starts in the first months of life. Occasionally the disorder extends to include the adjacent skin, adjacent scalp, and cheeks.
Ulerythema ophryogenes and keratosis pilaris have been described in patients with two similar but distinct “ RAS opathies” of the RAS-MAPK signaling pathway, the cardiofaciocutaneous (CFC) syndrome and Noonan syndrome. Patients with CFC syndrome often show widespread keratosis pilaris-like lesions of the face, ears, scalp, and extensor surfaces of the extremities that may be more lichenoid and prominent than keratosis pilaris. Sometimes patients with CFC and Noonan syndromes have alopecia of the eyelashes and eyebrows with follicular hyperkeratosis but lack the atrophy and scarring of ulerythema ophryogenes. Both CFC and Noonan syndromes share features of short stature, congenital cardiac abnormalities (particularly pulmonary valve stenosis), retardation, macrocephaly, hypertelorism, a high forehead, pectus carinatum, curly hair, and many pigmented nevi. Lymphedema and a low posterior hairline are more typical features of Noonan syndrome. Patients with CFC syndrome often show hypoplastic supraorbital ridges, bitemporal constriction, and an antimongoloid slant, features not described in Noonan syndrome.
As with clinical features, there is overlap in genes that are mutated in Noonan and CFC syndromes ( BRAF, KRAS, MEK1 , and MEK2 ). However, the genes most commonly altered in Noonan syndrome are PTPN11 (50%) and SOS1 (10% to 13%). Individuals with Noonan syndrome associated with loose anagen syndrome exclusively have a unique mutation in SHOC2, a scaffold protein required for the RAS-MAPK signaling cascade. A recent study noted genotype–phenotype correlation during the first year of life, with thin hair linked to mutations in SHOC2 and BRAF, whereas keratosis pilaris was associated with mutations in SOS1, BRAF, and SHOC2. Other RAS opathies with skin features are lentigines, electrocardiographic abnormalities, ocular hypertelorism, pulmonic stenosis, abnormal genitalia, retardation of growth, and sensorineural deafness (LEOPARD) syndrome; neurofibromatosis; neurofibromatosis-Noonan syndrome; Legius syndrome; the newly described CBL -mutation-associated syndrome (café-au-lait spots, see Chapter 11 ); and Costello syndrome (see Chapter 6 ). Ulerythema ophryogenes has also been associated with Cornelia de Lange and Rubenstein–Taybi syndromes, as well as with woolly hair (see Fig. 7-8 ).
Atrophoderma vermiculata (folliculitis ulerythema reticulata, atrophoderma vermicularis) usually has its onset between 5 and 12 years of age. This disorder is characterized by the formation of numerous tiny symmetric atrophic and at times erythematous pits on the cheeks, periauricular areas, and occasionally the forehead and eyebrows. These cribriform lesions generally measure 1 to 2 mm across and 1 mm deep and are separated from each other by narrow ridges of normal-appearing skin. Laser and dermabrasion have been advocated to improve the cosmetic appearance of affected individuals when the condition is stable, usually after puberty. Low-dose isotretinoin (0.5 mg/kg per day) for two 6-month courses has been reported to cause cosmetic improvement.
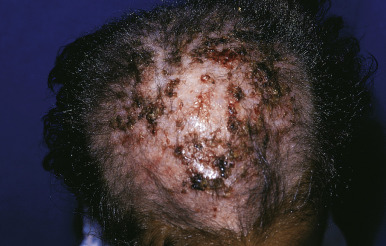
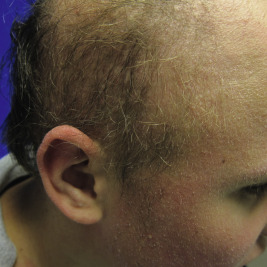
KFSD is characterized by atrophic keratotic follicular papules of the scalp, eyebrows, and eyelashes that eventuate in scarring alopecia ( Fig. 7-27 ). Associated features are palmoplantar keratoderma, corneal dystrophy, photophobia, and atopy. KFSD is usually an X-linked recessive disorder, and female carriers may show milder manifestations. The initial signs are photophobia with tearing, ophthalmitis, and conjunctival and corneal inflammation, which occur in the first weeks or months of life; congenital glaucoma and cataracts have been noted in association. Extensive keratosis pilaris of the face, extremities, and trunk tends to begin during early childhood, often in association with facial erythema. Cicatricial alopecia of the scalp begins around puberty and slowly progresses in association with follicular inflammation and fibrosis; eyebrows also tend to be affected. Some patients show palmoplantar keratoderma and marked xerosis. Acne keloidalis nuchae (see Acne Keloidalis section) and tufted hair folliculitis has been described in several patients. Ichthyosis follicularis, congenital atrichia, and photophobia (IFAP) is another X-linked condition in which affected neonates show keratotic follicular papules with a sandpapery feel to the skin, atrichia or severe hypotrichosis, and photophobia from birth ( Fig. 7-28 ). In contrast to KSFD, the alopecia of patients with IFAP does not scar. Mental retardation and developmental delay have been described in both KSFD and IFAP syndromes. Other features are gingival hyperplasia and angular stomatitis, psoriasiform plaques, palmoplantar erythema with thickening, short stature, and seizures.
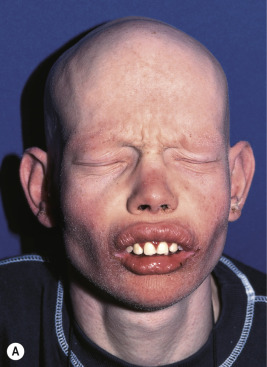
IFAP and KFSD are allelic and result from mutations in a zinc metalloprotease (MBTPS2) that is important for cholesterol homeostasis, handling endoplasmic reticulum stress, and cell differentiation. Mutations in MBTPS2 also cause X-linked Olmsted syndrome (see Chapter 5 ) and BRESEK/BRESHECK syndrome. The latter disorder is characterized by brain anomalies, retardation (intellectual, associated with microcephaly), ectodermal dysplasias (atrichia and photophobia, but not ichthyosis follicularis), skeletal deformities (especially vertebral and hand anomalies), Hirschsprung disease, eye or ear anomalies, cleft lip/palate or cryptorchidism, and kidney anomalies. The most common features are included in the BRESEK acronym.
KFSD may also be autosomal dominant in inheritance, although the underlying molecular defect is unclear. Marked facial erythema, extensive folliculitis, onychodystrophy, and multiple caries have been described in these patients.
Ichthyosis in association with hair abnormalities may also be seen in ichthyosis hypotrichosis syndrome, ichthyosis-hypotrichosis-sclerosing cholangitis syndrome, Netherton syndrome (see Chapter 5 ) and TTD. Another disorder with nonscarring partial alopecia that must be distinguished is hereditary mucoepithelial dysplasia. In addition to extensive keratosis pilaris and psoriasiform plaques, affected individuals show fiery red mucosal inflammation (hard palate, gingival, tongue, perianal, and perineal), and ocular photophobia with keratitis, cataracts, and corneal opacities.
The follicular scarring of these disorders should be distinguished from disorders of follicular atrophoderma without keratotic plugs. These include perifollicular atrophoderma of acne scarring (see Chapter 8 ), Conradi–Hünermann syndrome (see Chapter 5 ), Rombo syndrome, and Bazex syndrome. Treatment of these disorders of follicular plugging is challenging, with most patients refractory to systemic and topical steroids, systemic antibiotics, dapsone, methotrexate, and systemic retinoids.
Other Scarring Alopecias
Scarring or cicatricial alopecia is the end result of a wide number of inflammatory processes in and around the pilosebaceous units, resulting in irreversible destruction of tissue and consequent permanent scarring alopecia. The scarring may be the result of a developmental defect (aplasia cutis) (see Figs. 2-34 and 2-35 ); inflammatory changes due to severe bacterial, viral, or fungal infection; physical trauma (halo alopecia from caput succedaneum [see Chapter 2 ], irradiation, long-term trichotillomania, thermal or caustic burns); neoplastic or infiltrative disorders (including severe alopecia mucinosa); various dermatoses (lichen planus, lupus erythematosus, localized or systemic scleroderma [see Chapter 22 ]); keratosis pilaris atrophicans, a group of disorders of hair plugging; or various dermatologic syndromes such as folliculitis decalvans, dissecting cellulitis of the scalp, acne keloidalis, and pseudopelade.
Follicular Mucinosis
Follicular mucinosis (alopecia mucinosa) is an inflammatory disorder characterized by sharply defined follicular papules or infiltrated plaques with scaling, loss of hair, and accumulation of mucin in sebaceous glands and the outer root sheaths of affected hair follicles. A relatively uncommon condition affecting children as well as adults, the disorder is characterized by often pruritic, grouped follicular papules that often coalesce into scaling plaques or nodular boggy infiltrated plaques with overlying erythema and scaling ( Fig. 7-29 ). Lesions usually measure 2 to 5 cm in diameter. Distributed primarily on the face, scalp, neck, and shoulders (occasionally the trunk and extremities), lesions are usually devoid of hair. Except in the scalp or eyebrows, the alopecia is generally not conspicuous.
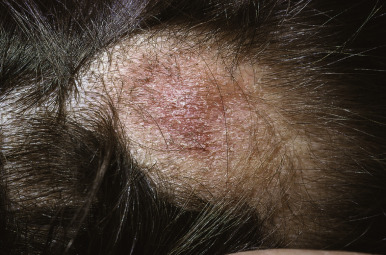
The cause of follicular mucinosis is unknown. In the majority of cases (in those <40 years of age), it is a benign idiopathic condition. In persons older than age 40, however, the presence of boggy infiltrated plaques of alopecia mucinosa may be the first sign of cutaneous T-cell lymphoma. Cutaneous T-cell lymphoma has rarely been described in children with follicular mucinosis, so affected children must be monitored carefully.
Follicular mucinosis must be differentiated from lichen spinulosus, pityriasis rubra pilaris, tinea infection, pityriasis alba, granulomatous diseases, and the papulosquamous group of disorders. When the diagnosis remains in doubt, cutaneous biopsy of an affected area is generally confirmatory, showing accumulation of mucin within the hair and sebaceous glands.
Solitary or few lesions usually clear spontaneously within 2 years. More numerous and widely distributed plaques tend to be more chronic. Destruction of follicles may give rise to permanent alopecia, and the disorder may persist with new lesions continuing to appear over years. Although some cases appear to benefit from topical or intralesional corticosteroids, such claims are difficult to evaluate because spontaneous healing is the rule.
Folliculitis Decalvans
Folliculitis decalvans is characterized by successive crops of patchy, painful folliculitis leading to progressive hair loss and scarring. The disorder must be distinguished by culture from bacterial folliculitis caused by Staphylococcus aureus, although the two forms often coexist, leading to the hypothesis that folliculitis decalvans results from an abnormal host response to staphylococcal toxins. Folliculitis decalvans may begin during adolescence in male patients but is rarely seen in female patients before 30 years of age. Although the scalp is the most commonly affected site, hair-bearing areas of the trunk, axillae, and pubic region may be affected. Typical lesions show round to irregular bald, atrophic patches, each surrounded by crops of follicular pustules. Tufted folliculitis is a variant in which tufts of hair emerge from dilated follicular openings amid areas of scarring.
Treatment of folliculitis decalvans is difficult. Systemic antibiotics that penetrate the follicle well (tetracyclines, clindamycin, erythromycin) with or without rifampicin often prevent disease extension, but continued administration is required to prevent relapse. Many individuals develop severe alopecia and scarring despite long-term and intensive therapy.
Dissecting Cellulitis of the Scalp
Dissecting cellulitis of the scalp, also termed perifolliculitis capitis abscedens et suffodiens, is characterized by painful fluctuant nodules and abscesses of the scalp connected by tortuous ridges or deep sinus tracts with cicatricial alopecia. The connection can often be demonstrated by applying pressure to one nodule and observing purulent drainage emerging from another. Lesions are usually first noted at the occipital area or vertex but may progress to involve the entire scalp. The disorder occurs most commonly in African-American male teenagers and young adults. An association with acne conglobata and hidradenitis suppurativa has been described (“follicular occlusion triad”), suggesting an inflammatory reaction to Propionibacterium acnes . Other pustular disorders of the scalp, including bacterial folliculitis and inflammatory tinea capitis (kerion), must be considered.
The disorder has a chronic, relapsing course. Although use of oral tetracyclines or erythromycin with or without surgical drainage of lesions may be effective, many studies suggest systemic administration of isotretinoin to be the treatment of choice. Laser ablation has also been advocated.
Acne Keloidalis
Acne keloidalis (folliculitis keloidalis) is a chronic scarring folliculitis and perifolliculitis of the nape and occipital scalp. Initial lesions tend to be inflammatory papules and occasionally pustules that evolve into firm keloidal, often coalescent papules and plaques ( Fig. 7-30 ). Severe cases may show abscesses and sinus formation. Patients may complain of pruritus and discomfort. The disorder is seen most commonly in postpubertal males, especially African-Americans between the ages of 14 and 25 years, but is occasionally described in females.
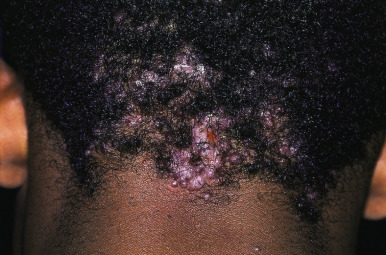
Acute inflammation of the follicle is thought to be the primary pathologic process, followed by a granulomatous foreign body reaction to released hair, and subsequent fibrosis; a variety of triggers have been proposed, among them irritation from shirt collars or helmets, bacterial folliculitis, or ingrown hairs after a short haircut. Treatment of this disorder is difficult and consists of long-term systemic antibiotics and intralesional corticosteroids. Patients should avoid close “clipper” haircuts and scratching of the area. Laser and excision with second-intention healing have been helpful for selected patients, especially with fibrotic nodules. Targeted ultraviolet B (UVB) light therapy has shown promise.
Pseudofolliculitis Barbae
Pseudofolliculitis barbae, commonly called razor bumps or ingrown hair, is an inflammatory condition of hair follicles of the beard area that is particularly common in adolescent males of African ancestry with curly, coarse hair (see Chapter 14 ). The condition can affect adolescent girls with tightly curled hair, however, especially if shaving occurs on the face because of hirsutism or because of hair removal elsewhere (waxing plucking or shaving of the axillae or pubic area). Erythematous, 2- to 4-mm, flesh-colored or often hyperpigmented, follicular-based papules are characteristic. The shape of the hair follicle, hair cuticle, and direction of hair growth predispose the patient to the inflammatory response when hair is shaved or plucked. A single-nucleotide polymorphism (SNP) in KRT75, encoding hair keratin 75, has been linked with a much greater tendency to develop pseudofolliculitis barbae, especially in individuals with curly hair. It is theorized that the pressure and traction exerted by close and regular shaving destabilizes the hair keratin.
Pseudofolliculitis barbae is thought to represent a foreign-body reaction around an ingrown hair. Therapy largely involves prevention, particularly by temporarily discontinuing shaving or other forms of hair removal and then instituting alternative techniques that decrease the closeness of the shave such as use of an electric razor (avoiding the “closest” shave setting), the Bumpfighter razor (American Safety Razor Company, Staunton, VA), or chemical depilatories. The condition tends to clear 4 to 8 weeks after discontinuation of triggers. Shaving should always be in the direction of hair growth, and pretreatment with an antibacterial soap or benzoyl peroxide wash may decrease the potential inflammation from bacterial overgrowth. Adjunctive topical agents are retinoids, low-potency topical steroids, topical antibiotics, and depigmenting agents. Nonlaser epilation is not recommended and may exacerbate pseudofolliculitis barbae; however, the neodymium:yttrium-aluminum-garnet (Nd:YAG) 1064 nm and diode (800 to 810 nm) lasers have been used to epilate the curly hairs and can cause significant improvement. Eflornithine cream, which inhibits hair growth, may also be helpful but may be associated with the development of local irritation.
Pseudopelade
Pseudopelade is a nonspecific scarring form of slowly progressive alopecia of the scalp generally seen in adults, although it has been described rarely in children. It may represent the end result of discoid lupus erythematosus (see Chapter 22 ) or lichen planopilaris (see Chapter 4 ). It is characterized by multiple small round, oval, or irregularly shaped hairless cicatricial patches of varying sizes. Affected areas are shiny, ivory white or slightly pink, and atrophic. Lesions commonly coalesce to form finger-like projections and have been compared to footprints in the snow. Lesions generally appear at the vertex of the scalp. A few hair-containing dilated hair follicles may be interspersed between the patches. The condition tends to resolve spontaneously after several years, leaving the alopecia. Therapy is often unsuccessful, although intralesional injections of triamcinolone have temporarily benefited some patients. Cosmetic improvement has also been achieved by the multiple-punch autograft technique of hair transplantation.
Telogen Effluvium
The normal cyclic pattern of anagen and telogen hair phases may be interrupted by a variety of different stimuli, resulting in telogen effluvium. Telogen effluvium represents the most common type of alopecia in children and is characterized by diffuse thinning of scalp hair to varying degrees ( Fig. 7-31 ). The average individual who shampoos at least every other day loses 50 to 100 telogen hairs per day, but 25% of scalp hair (25,000 hairs) must be shed before unmistakable thinning becomes apparent.
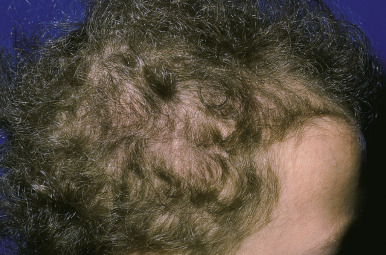
Several stimuli are capable of producing an interruption in the anagen phase of the hair follicles ( Box 7-5 ). Telogen effluvium may be suggested by a history of a stressful event preceding the onset of alopecia by 6 to 16 weeks that shifts more hairs into telogen phase. Most common are acute illnesses, especially with fever, major trauma, surgery, or childbirth. Initiation of medications ( Box 7-6 ) or discontinuation of medications, particularly oral contraceptives, isotretinoin, anticonvulsants, cimetidine, and terbinafine, has also been implicated. More chronic telogen effluvium has been associated with chronic illness, thyroid abnormalities, iron deficiency anemia, malabsorption (e.g., celiac disease), malnutrition (e.g., anorexia nervosa), systemic lupus erythematosus, and zinc deficiency. The proportion of follicles affected and the severity of the subsequent alopecia depend on the duration and severity of the trigger and individual variations in susceptibility.
Emotional stress
Fever (high)
Medications (see Box 7-6 )
Nutritional disorders
Biotin deficiency
Dieting, crash; anorexia
Essential fatty acid deficiency
Iron deficiency
Hypervitaminosis A
Physiologic telogen effluvium of the newborn
Parturition
Severe chronic illness
Severe infection
Surgery
Thyroid disease (hyperthyroidism or hypothyroidism)
Albendazole
Amphetamines
Angiotensin converting enzyme inhibitors (e.g., captopril, enalapril)
Anticoagulants (e.g., heparin, warfarin)
Anticonvulsants (e.g., valproic acid, carbamazepine)
β-Blockers (e.g., propranolol)
Cimetidine
Danazol
Interferon-α
Lithium
Oral contraceptions (during use or with discontinuation)
Retinoids (e.g., isotretinoin)
The diagnosis may be confirmed by both counting the number of hairs shed each day and determining the percentage of telogen hairs in the scalp. Telogen hair represents approximately 15% and anagen 85% of scalp hair. If 25% or more of gently pulled hairs are telogen, telogen effluvium can be diagnosed. Anagen hair roots can be recognized by their intact outer and inner hair sheaths, with or without a portion of the dermal papilla adherent to the tip of the root (see Fig. 7-1, A ). Telogen hair roots have uniform shaft diameters, contain no pigment, and are club shaped (see Fig. 7-1, B ), much like the tip of a cotton-tipped applicator.
Increased telogen hair loss can also be seen in children with short anagen syndrome, in which the short, fine hair does not require haircuts and is present from birth. The shortening of the anagen phase, despite a normal rate of growth, leads to the decrease in the maximal hair length and an increase in the number of hairs in telogen. The disorder tends to resolve spontaneously during puberty and adulthood. Elongation of the anagen phase has also recently been described as a result of mutations that deplete fibroblast growth factor 5 (FGF5), a regulator of hair length. Because anagen hairs are already the majority of scalp hair but represent the minority of hairs of the eyelashes and extremities, trichomegaly (very long eyelashes) and longer extremity hair are the clinical manifestations in this recessive disorder that is homologous to the “angora” mutation in several species.
There is no effective treatment for telogen effluvium, but complete regrowth almost invariably occurs within months unless the stressful event is repeated or the underlying trigger is sustained. Careful explanation of the cause of this disorder and its favorable prognosis, with careful instructions to the patient to avoid unnecessary manipulation, tends to suffice. Blood tests for underling disorders should be performed based on personal and family history and on examination, especially if no trigger is obvious. Adolescents predisposed to AGA may show incomplete regrowth after telogen effluvium with a residual pattern of hair loss consistent with AGA. Rarely, prolonged illness with high fevers destroys some follicles completely and only partial recovery ensues. Telogen effluvium occasionally occurs more than once in an individual, suggesting a predisposition to more significant hair loss with stress.
Anagen Effluvium
In anagen effluvium, hair shaft production is markedly reduced, leading to tapering of the shaft and shedding. Given that more than 80% of scalp hair is in anagen phase, hair loss is usually profound. Anagen effluvium usually occurs in patients administered radiation or chemotherapy for malignancy. Most commonly implicated are cyclophosphamide, methotrexate, 6-mercaptopurine, and doxorubicin. In addition, anagen effluvium may be associated with exposure to colchicine and toxic levels of boric acid, lead, thallium, arsenic, bismuth, and warfarin.
The clinical features of anagen effluvium depend on the degree of toxicity created by the causative agent. With lower doses of the toxic agent, only segmental thinning or narrowing may occur without actual fracture of the hair shaft. Gentle hair pulls yield “pencil point” dystrophic hairs with proximal tips tapered to a point. With extensive anagen effluvium, the remaining hairs are telogen, and the hair plucks late in the course may show as many as 100% telogen hairs. A careful history, documented evidence of hair loss, microscopic examination of spontaneously shed and manually epilated hairs, and appropriate physical and toxicologic examinations help to establish the correct diagnosis. Cessation of the responsible drug or toxin generally results in regrowth of hair.
Alopecia Areata
Alopecia areata (AA), one of the most common hair loss disorders with a prevalence of 0.1% to 0.2% of the population, is characterized by the sudden appearance of sharply defined round or oval patches of hair loss. Alopecia totalis (loss of all scalp hair) or alopecia universalis (loss of all body hair) develops in approximately 5% of cases. The cumulative lifetime risk of developing AA is 2.1%, with the peak incidence occurring during young adulthood. Prepubertal onset occurs less often than onset during adolescence but is not uncommon and is associated with a family history of AA and a poorer prognosis. Occurrence in young infants is unusual, and the disorder is very rare in neonates.
There is a strong association between AA and atopic dermatitis, which is greatest among prepubertal children and in individuals with alopecia totalis and alopecia universalis. Loss-of-function mutations in FLG (encoding filaggrin) have been associated with a more severe course and associated atopy but not a higher risk. There is no sexual predilection for the disorder. Familial occurrence is reported in 8% to 52% of children, with an estimated lifetime risk of 7.1% in siblings, 7.8% in parents, and 5.7% in offspring. Occurrence in both identical twins is 55%, emphasizing the importance of genetic as well as environmental factors.
The typical clinical picture of AA generally consists of a sudden (overnight or several days) appearance of one or more round or oval, well-circumscribed, clearly defined patches of hair loss ( Fig. 7-32 ). Overall 83% of children have this patchy loss involving less than 50% of the scalp. Occasionally the initial patches may lack a regular outline and at times may demonstrate scattered long hairs within the bald areas. In other instances the initial loss may be diffuse, with discrete patches of alopecia being apparent only after 1 or 2 weeks, if at all. The primary patch may appear on any hairy cutaneous surface but usually occurs on the scalp. The skin is smooth, soft, and almost totally devoid of hair. Rarely, slight erythema or edema may be found at an early stage. Depigmented or hypopigmented hair shafts, simulating poliosis, may be seen ( Fig. 7-33 ). Discrete islands of hair loss sometimes are separated by completely uninvolved or partially involved scalp. Around the margins of patches of alopecia, pathognomonic “exclamation-mark” hairs may be detected ( Fig. 7-34 ). These loose hairs, with attenuated bulbs and short stumps, are easily plucked out of the scalp. Examination of such hairs under a low-power microscope reveals an irregularity in diameter and a poorly pigmented hair shaft that tapers to an attenuated bulb. The hair bulb represents the dot of the exclamation point. Dermoscopic evaluation of affected scalp can be very helpful, showing the tapered hairs and sometimes yellow perifollicular dots, demarcating the hyperkeratotic follicular plugs.
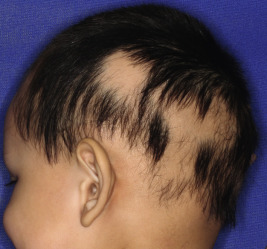
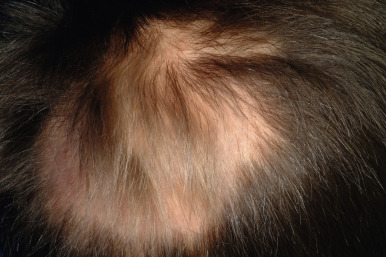
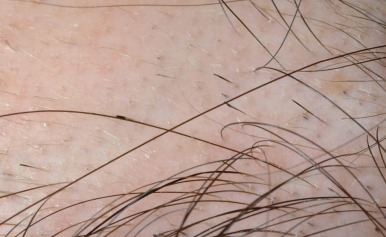
The ophiasis pattern of hair loss begins as a bald spot on the posterior occiput and extends anteriorly and bilaterally in a 1- to 2-inch wide band encircling the posterior scalp, usually extending above the ear but occasionally to the anterior aspect of the scalp ( Fig. 7-35 ). The ophiasis pattern is generally associated with a poor prognosis. Progression to the totalis or universalis forms ( Fig. 7-36 ) occurs more slowly but more often in children than in adults. Alopecia may involve any hair; eyebrows and eyelashes may be lost with or without patches of hair loss on the scalp ( Fig. 7-37 ).
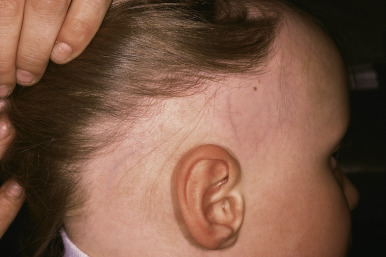
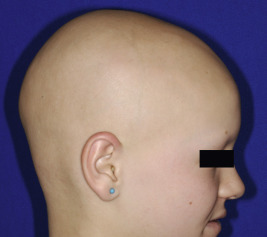
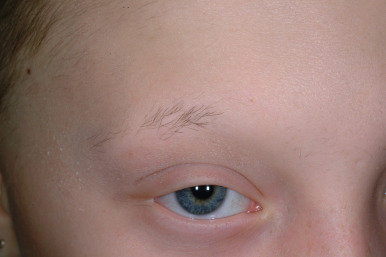
Nail defects are seen in 10% to 20% of cases. Although more extensive disease is associated with more nail involvement, some patients may have extensive nail dystrophy with little hair change. The most characteristic nail abnormality is a fine, grid-like stippling, regularly arranged in horizontal and/or vertical rows with smaller and shallower pits than those seen in patients with psoriasis ( Fig. 7-38, A ). Proximal shedding (onychomadesis), opacification, and serration of the free edges may also be seen ( Fig. 7-38, B ). Longitudinal ridging (trachyonychia) is another associated finding. Nail dystrophy tends to occur more often and be more severe in patients with alopecia totalis and universalis.
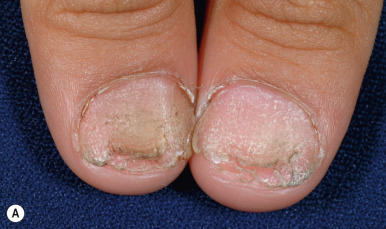
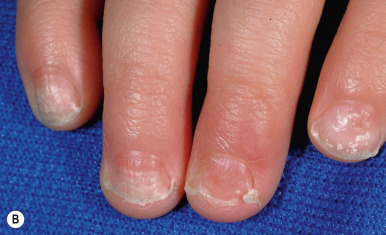
The diagnosis of AA is based on its clinical picture. The sudden appearance and circumscribed nonscarring, patterned nature of hair loss distinguish it from other disorders of alopecia. Trichotillomania is typically associated with bizarre, irregular patches of hair loss with areas of broken hairs of different lengths. The absence of signs of inflammation and scaling will generally help distinguish this disorder from that of tinea capitis. When the diagnosis is in doubt, microscopic examination of hairs, potassium hydroxide mounts, fungal cultures, and cutaneous punch biopsy will generally establish the proper diagnosis. Alopecia univeralis beginning in infants must be distinguished from congenital alopecias such as atrichia with papules. The AA spectrum can be a feature in 33% of children with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED, see Chapter 23 ) syndrome because of mutations in AIRE . Although associated autoimmune disorders in affected children are quite rare, a family history of other autoimmune disorders, especially thyroiditis, is common, and autoantibodies may be detected in patient sera. An increased incidence has also been noted in patients with trisomy 21.
The course of AA is variable and difficult to predict. New patches of hair loss may appear for 4 to 6 weeks and occasionally for several months. Spontaneous regrowth may occur. In general, when the process is limited to a few patches, the prognosis is good, with complete regrowth occurring within 1 year in 60% to 80% of patients; progression to total loss of scalp hair is unusual (<10%) and portends a considerably worse prognosis. About 30% of patients with patchy AA will have future episodes once regrown. In general, the earlier the onset, the poorer the prognosis. Other prognostic indicators of a worse outcome are family history of autoimmune disease, personal history of atopy, and nail abnormalities. Therapy for AA at best controls the condition but does not cure it or prevent development of new areas. Because of the chronic nature of the condition and the slow growth of hair, any trial of a treatment modality requires at least 4 months. The sudden hair loss, cosmetic ramification, and unpredictable course make AA a frightening disorder for affected patients and families ; psychological support and counseling are required for all patients and parents. Adequate camouflage of alopecic areas may be achieved by hats, headbands, or hairstyle changes. In children with severe involvement, wigs can be helpful. Locks of Love is an organization that provides hair prostheses to financially disadvantaged children under the age of 18 years ( www.locksoflove.org ). The National Alopecia Areata Foundation ( www.alopeciaareata.com ) is a national support group for affected children and their families. Given the high percentage of children with patchy AA who show spontaneous remission within a year, some physicians prescribe no therapy for this condition. In general, patients with more extensive AA or with limited involvement unresponsive to topical corticosteroids should be referred to a dermatologist.
The most commonly used therapy for more limited AA in children is topical corticosteroids, with or without occlusion (such as may be achieved under a wig, bathing cap, or Saran Wrap); class II (potent) steroids are usually administered. Because the regrowth rate without medications is high, it has been hard to demonstrate clear evidence of the efficacy of steroids. If a class I (ultrapotent) steroid is employed, its use should be limited to intermittent pulse therapy; longer use may result in significant local atrophy and systemic absorption. In one study, clobetasol cream was more effective than 1% hydrocortisone in decreasing extent of hair loss by 12 weeks of twice daily use when used for 6 weeks on and 6 weeks off for a total of 24 weeks. Intradermal corticosteroid injections commonly result in regrowth in tufts at injection sites within 4 to 6 weeks but are too uncomfortable for most children, even with the use of topical anesthetic creams. When an intralesional corticosteroid is used, a syringe with a 30-gauge needle or jet injection is best. Triamcinolone acetonide is injected in concentrations of 2.5 mg/mL (eyebrow area) to 10 mg/mL (scalp). Dosage should be limited to 0.1 mL per site, spaced at least 1 cm apart, with injections at intervals of at least 4 to 6 weeks. Transient local atrophy may occur. Efficacy appears to be greatest in those who have less than 75% hair loss and with a relatively short duration of hair loss.
Minoxidil has been shown to stimulate follicular DNA synthesis. Although largely used for AGA, 2% to 5% topical minoxidil solution has been shown to cause cosmetically acceptable hair regrowth in approximately 20% to 45% of patients when used twice daily for 2 months. Best results occur in patients with limited hair loss and when used concurrently with topical corticosteroids or anthralin. Cutaneous side effects may include local irritation, allergic contact dermatitis, and hypertrichosis, especially on the forehead ( Fig. 7-39 ). Although quite rare, three children treated with topical minoxidil developed tachycardia, palpitations, and dizziness. Prostaglandin F2a analogues (e.g., latanoprost, bimatoprost, travoprost) have not been helpful for alopecia of the eyelashes or brows and have been linked to periorbital hyperpigmentation.
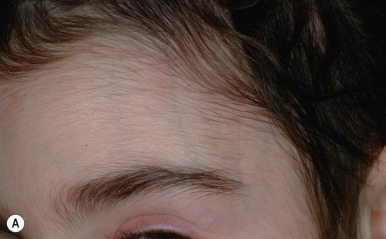
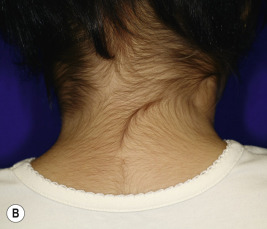
Anthralin cream is an alternative therapy that seems to elicit hair growth by nonspecific immunostimulation. The 1% cream is usually applied as short-contact therapy, initially for 30 minutes with a gradual increase in exposure as tolerated to a maximum of 2 hours before shampooing of the scalp. A mild dermatitis is often required for regrowth, so concurrent use of topical corticosteroids is not recommended. New hair growth is usually seen in 3 months, but 6 months or more may be required for an acceptable response. Scalp irritation, folliculitis, and staining of the skin or clothes are potential adverse effects.
Two studies have demonstrated significantly low levels of vitamin D in individuals with AA, and a boy with AA responded to topical calcipotriol, but no causal relationship with vitamin D deficiency has been confirmed. Excimer laser therapy twice weekly is painless and caused hair regrowth in 60% of recalcitrant patches of AA in children in less than 3 months; only 22% of responders relapsed after 6 months.
Treatment of more extensive AA (>50%), alopecia totalis, and alopecia universalis is very difficult. Topical application of corticosteroids is generally not helpful, but anthralin has shown greater responses. Topical immunotherapy is considered the treatment of choice for chronic, severe AA and has been shown to decrease the T-cell infiltration. Patients are initially sensitized to the contact allergen (2%); then increasing concentrations of the allergen beginning at 0.001% (occasionally stronger) are applied to the scalp until mild erythema and scaling develop. Squaric acid dibutyl ester and diphenylcyclopropenone are used most commonly, because they are effective but not mutagenic or carcinogenic. Contact dermatitis at the sensitization site (about 20% of treated children) or treatment site and regional lymphadenopathy are the risks of this therapy but only occasionally lead to discontinuation. Immunotherapy may be performed weekly by the physician or administered with careful monitoring by responsible parents at home on a daily basis, gradually increasing the frequency of application and dosing. Up to 40% respond to immunotherapy with complete regrowth, although 70% to 80% show some response. Unfortunately, relapse occurs in 81% of responders. In patients who are atopic with extensive AA, the addition of oral fexofenadine to the contact allergen treatment may increase the response. Although photochemotherapy with systemic psoralen followed by ultraviolet A (PUVA) light and cyclosporine therapy have been successful in some adults with this disorder, their efficacy and potential toxicity probably do not justify their use in children.
Although not recommended for general use, systemic corticosteroids may be considered for carefully selected patients with severe involvement and rapidly progressive hair loss who are psychologically handicapped by their disorder. In such instances, prednisone may be administered in dosages of 0.5 to 1 mg/kg per day for 4 weeks until hair loss ceases and then tapered to alternate-day therapy for a few months. It must be emphasized that close follow-up evaluation is indicated in such cases and that the potential side effects associated with systemic corticosteroid therapy must be explained to the patient and parents. Pulse therapy with intravenous methylprednisolone has also been administered at a dosage of 250 mg twice daily for three sequential days with cessation of hair loss, but the long-term outcome after corticosteroid therapy is poor. In one retrospective study of 24 children with multifocal disease to alopecia universalis, 5 to 6 monthly treatments on 3 consecutive days of 8 mg/kg intravenous methylprednisolone led to a complete response in 38% and no response in 33%, but 81% relapsed within a mean of 9.5 months after therapy. Not surprisingly, patients with duration of less than 6 months and an onset at younger than 10 years of age and multifocal vs. diffuse disease responded better. Continuing application of topical minoxidil after a steroid taper has been shown to decrease hair loss. Methotrexate (0.3 to 0.6 mg/kg per week) led to greater than 50% hair regrowth in 5 of 12 children (>40%) with recalcitrant AA in whom it was used for more than 2 months. The mean time to regrowth was 4.4 months, and all responders experienced improvement within 6 months. Sulfasalazine has shown cosmetically acceptable improvement in about 25% of treated adults when used at a dosage of 1.5 g twice daily (starting instead with 0.5 g/dose and then 1 g/dose for the first and second month, respectively). Trials with TNF inhibitors have been disappointing, and AA has developed during TNF inhibitor therapy.
Recent studies, including a genome-wide association study (GWAS) of more than 1000 affected individuals, have shed light on the underlying pathomechanism of AA. Whereas most of the linked genes involve the immune system, a receptor for stress-induced proteins ( NKG2D ) is also strongly linked, which is of interest because a stressful event precedes the onset of AA in 9.5% to 58% of patients. These mechanistic studies have led to the discovery that systemic administration of Janus kinase (JAK) inhibitors (ruxolitinib, tofacitinib) prevent the development of AA in the C3H/HeJ -grafted mouse model and in pilot studies lead to almost complete hair regrowth after up to 5 months of 20 mg ruxolitinib twice a day orally in adults with severe AA. Topical administration of ruxolitinib and tofacitinib to established mouse AA also led to full hair regrowth within 12 weeks, suggesting the applicability of topical therapy for pediatric AA.
Androgenetic Alopecia
Androgenetic alopecia (AGA) occurs in both males (common balding or male-pattern baldness) and females (hereditary thinning or female-pattern hair loss) and is the most common cause of hair loss in adolescents and adults. In many cases, it begins in teenage years with onset as young as 7 years ; in general, the earlier the onset, the more profound the subsequent alopecia. The disorder is characterized by patterned, progressive hair loss from the scalp and results from the effects of circulating androgens in genetically susceptible individuals. Dihydrotestosterone is the primary androgen implicated, converted from testosterone by the enzyme 5-α-reductase. These androgens gradually decrease the size of scalp hair follicles, resulting in miniaturized hairs. In addition, the anagen growth phase is shortened, leading to more hairs in telogen phase. The condition is inherited as a polygenic trait influenced by both maternal and paternal genes ; in one study, 83% of adolescents with AGA had an affected first-degree relative.
Most patients with AGA note thinning of scalp hair rather than shedding, although shedding may occur early in the course and be confused with telogen effluvium or diffuse AA. AGA during adolescence occurs twice as often in males as in females and is generally more severe. The mildest and often earliest form of androgenetic male-pattern alopecia in males is thinning at the vertex, often in conjunction with symmetrical bitemporal triangular recession of the hairline ( Fig. 7-40 ). In girls, the frontal hairline is relatively unaffected and there is either just vertex thinning or more commonly, diffuse thinning, particularly from the frontal scalp to the vertex (see Fig. 7-38 ). Widening of the central hair part is often seen, leading to scalp visibility ( Fig. 7-41 ). This female pattern of more diffuse hair loss is observed in 20% to 33% of adolescent males.