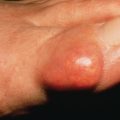
An inflammatory disorder that is primarily localized in the subcutaneous fat is termed a panniculitis. This group of disorders may be challenging for both the clinician and the dermatopathologist. Clinically, in all forms of panniculitis, lesions present as subcutaneous nodules. Histopathologically, the subcutaneous fat is a rather homogeneous tissue, and inflammatory processes may show considerable overlap. One way of classifying panniculitis is to separate them into those that involve the septae between fat lobules (septal panniculitides) from those processes that primarily involve the fat lobules (lobular panniculitides). The prototypical septal panniculitis is erythema nodosum. Some lobular panniculitides are caused by vasculitis (e.g., polyarteritis nodosa) and are discussed in other chapters. The remaining lobular panniculitides are categorized by their pathogenesis. Weber-Christian disease, Rothmann-Makai disease, lipomembranous or membranocystic panniculitis, and eosinophilic panniculitis are reaction patterns and are not specific entities. Neutrophilic panniculitis may be infectious or may represent a variant of Sweet syndrome with primary involvement of the panniculus.
Given the depth of lesions in the panniculus, the choice of biopsy is critical in establishing the diagnosis. An incisional or excisional biopsy, narrow at the skin surface and wider in the panniculus, is the optimal procedure. An alternative double-punch method, using a 6–8 mm punch first, followed by a 4–6 mm punch at the depth of the first punch, may be considered, but it is less ideal. Depending on the amount of fat in the particular body location that is being biopsied and the age of the patient, the fat can be adequately sampled with the initial punch. Panniculitis is an area of dermatopathology where the skill of the dermatopathologist is critical in establishing good clinicopathologic correlation. If the biopsy report from an adequate specimen does not match the clinical findings, the clinician should repeat the biopsy or ask for a second opinion on the original specimen.
Requena L: Normal subcutaneous fat, necrosis of adipocytes and classification of the panniculitides. Semin Cutan Med Surg 2007; 26: 66.
Zelger B: Panniculitides, an algorithmic approach. G Ital Dermatol Venereol 2013; 148: 351.
Septal Panniculitis (Acute and Chronic Erythema Nodosum)
Erythema nodosum (EN) is the most common inflammatory panniculitis. It occurs in two forms: acute, which is more common, and chronic, which is rare. Acute EN may occur at any age and in both genders, but most cases occur in young adult women (female/male ratio, 3 : 1–6 : 1). The eruption consists of bilateral, symmetric, deep, tender nodules and plaques 1–10 cm in diameter. Usually, there are up to 10 lesions, but in severe cases many more may be found. Initially, the skin over the nodules is red, smooth, slightly elevated, and shiny ( Fig. 23.1 ). The most common location is the pretibial area and lateral shins.
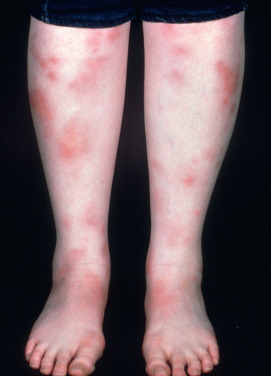
In general, the lesions should be primarily anterior rather than posterior calf. Lesions may also be seen on the upper legs, extensor arms, neck, and rarely the face. The onset is acute and is frequently preceded by malaise and leg edema. Arthritis or arthralgia, usually of the ankles, knees, or wrists, can occur. Fever, headache, episcleritis, conjunctivitis, and various gastrointestinal (GI) complaints may also be present. EN is a panniculitis that is a reactive response to another disease, therefore some of these symptoms may be related to the associated disease. Over a few days, the lesions flatten, leaving a purple or blue-green color resembling a deep bruise (erythema contusiforme). Ulceration does not occur, and the lesions resolve without atrophy or scarring. The natural history is for the nodules to last a few days or weeks, appearing in crops, and then slowly involute. EN in children affects boys and girls equally.
EN is a reactive process although approximately half of cases are idiopathic. It is frequently associated with a streptococcal infection (especially in children). Tuberculosis (TB) remains an important cause in areas where TB is endemic. Intestinal infection with Yersinia, Salmonella, or Shigella may precipitate EN. Other infectious causes include systemic fungal infections (coccidioidomycosis, histoplasmosis, sporotrichosis, blastomycosis) and toxoplasmosis. EN-like lesions have been described in other infectious diseases such as Helicobacter septicemia, brucellosis, psittacosis, and cat-scratch fever. Because these organisms are fastidious, it has not always been possible to exclude the possibility that the EN-like lesions seen in these diseases actually represent septic foci in the fat.
Sarcoidosis may present with fever, cough, joint pains, hilar adenopathy, and EN. This symptom complex, known as Löfgren syndrome, is especially common in Scandinavian, Irish, and Puerto Rican women. Sarcoidosis associated EN has been linked to a tumor necrosis factor–α (TNF-α) polymorphism. It generally responds well to therapy and runs a self-limited course. EN is frequently seen in patients with inflammatory bowel disease (IBD), more often Crohn disease than ulcerative colitis, and has been linked to mutations in genes controlling immune signaling, including PTGER4, ITGAL , and IKZF1 . In IBD patients, EN is not associated with overall disease severity but is strongly associated with female gender, eye and joint involvement, and isolated colonic involvement. Fecal calprotectin is a marker of inflammation in the GI tract and can be a sensitive marker for active IBD and thus a useful screening test. EN has been rarely reported in association with various hematologic malignancies, but Sweet syndrome and pyoderma gangrenosum are more commonly neutrophilic dermatoses associated with hematologic malignancy. EN has also been associated with idiopathic granulomatous mastitis, which is usually unilateral and mimics infection and breast carcinoma.
Drugs may also induce EN. The bromides and iodides were once the most frequent causative agents. Currently, oral contraceptives, hormone replacement therapy, sulfonamides, and penicillins are the most common medications inducing EN. Azathioprine has also been implicated in causing neutrophilic dermatoses including EN. Other newer associated medications include lenalidomide, methimazole, and Echinacea. The association with hormonal-based therapy, predominantly in young women, and the occurrence of EN in pregnancy suggest that estrogens may predispose to the development of EN. BRAF targeting therapy with vemurafenib, dabrafenib, or trametinib may also induce EN. Although infliximab has been used to treat EN associated with Crohn disease, it has also produced EN on multiple challenges in the setting of ankylosing spondylitis.
Erythema nodosum–like lesions have been described in Behçet syndrome and Sweet syndrome and probably represent these inflammatory processes occurring in the fat, rather than the coexistence of two disorders. Histologically, the subcutaneous lesions of Behçet syndrome show features different from EN: a lobular or mixed lobular and septal pattern and, most important, a vasculitis that may be lymphocytic or leukocytoclastic or that may involve a small arteriole. This vasculitis is proposed to be the primary event producing the subcutaneous lesions in Behçet syndrome.
A more chronic variant of EN, called chronic EN, erythema nodosum migrans, or subacute migratory panniculitis of Vilanova and Piñol, is well described. This form of septal panniculitis is much less common than acute EN. It is distinguished from acute EN because it is unilateral, or asymmetric if bilateral ( Fig. 23.2 ); it tends to occur in older women; and it is not associated with associated systemic symptoms except arthralgias. Additionally, the lesions in chronic EN begin as a single red nodule that tends to resolve but migrates centrifugally, forming annular plaques of subcutaneous nodules with central clearing. The lesions are painless or less tender than acute EN, and they have a prolonged course of months to years. TNF-α antagonists seem to be helpful in some cases.
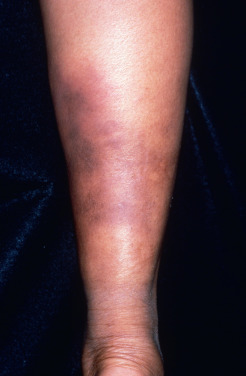
In the differential diagnosis of EN, other forms of panniculitis must be considered. Erythema induratum is a reactive process due to Mycobacterium tuberculosis. It usually affects primarily the posterior calves alone and runs a more chronic course, with the possibility of ulceration and scarring. Syphilitic gummas, as well as the nodules of sporotrichosis, are generally unilateral. Subcutaneous fat necrosis associated with pancreatitis and nodular vasculitis may also occur on the shins, but associated clinicohistologic features allow the differentiation from EN. Subacute infectious processes, such as Helicobacter cellulitis and primary atypical mycobacterial infection, may closely mimic EN. In most cases, the classic picture of the acute onset of symmetric, red, tender nodules on the anterior shins of a young woman readily leads to the diagnosis of EN without a biopsy. However, if the case is atypical or does not evolve typically, a biopsy should be performed. When the diagnosis of EN has been made in error, either the clinical features were atypical and a biopsy was not performed or was inadequate (punch biopsy), or the biopsy was misinterpreted by the pathologist.
The histopathology of EN is a septal panniculitis; the inflammatory infiltrate principally involves the connective tissue septa between fat lobules throughout the evolution of the lesion. The infiltrate may be composed of either neutrophils (early) or lymphocytes and other mononuclear cells (later), or a mixture, depending on the stage at which the lesion is biopsied. In older lesions, histiocytes and multinucleate giant cells may predominate. Fat lobules are only secondarily affected by the inflammation, but some foamy histiocytes may be seen in the evolution of the lesions. Meischer radial granulomas, which are aggregates of histiocytes around stellate clefts, are characteristic but not diagnostic of EN. Leukocytoclastic vasculitis is not a histologic feature of EN. In chronic EN, septal fibrosis and septal granulomas composed of epithelioid histiocytes are seen.
The management of EN involves three components: identifying the trigger, rest and elevation of the affected extremities, and antiinflammatory medications. Because streptococcus is a common trigger, throat culture (or perianal culture in young children) and antistreptolysin O (ASO) titer are indicated, especially in children. Some basic laboratory testing is done to rule out pregnancy in women, and stool calprotectin level is determined to evaluate for IBD. A complete history of any preceding illness will often lead to clues; for example, previous diarrhea might suggest Yersinia infection and a stool culture can be helpful if warranted based on symptoms. A travel and exposure history is especially important when considering endemic fungal infections. Because 4% of patients with histoplasmosis present with EN, this cause should be excluded in endemic areas, and TB should be excluded in patients who may have had exposure.
Early treatment of the infectious cause does not appear to shorten the duration of the EN, although EN triggered by infections tends to last longer with a more chronic infection, and streptococcal-induced EN tends to be shorter than TB-triggered EN. Bed rest is of great value and may be all that is required in mild cases, especially in children. Gentle support hose are also helpful. Curtailing vigorous exercise during the acute attacks will shorten the course, and restriction of physical activities might prevent exacerbations and recurrences. Aspirin and nonsteroidal antiinflammatory drugs (NSAIDs) such as indomethacin are often helpful. Potassium iodide is a safe and effective treatment for both acute and chronic EN. As a supersaturated solution, 5 drops three times a day, increased by 1 drop per dose per day up to 30 drops three times a day, is one easy-to-remember dose schedule. As a tablet, the dose is one 300-mg tablet three times daily. Induction of hypothyroidism by prolonged iodide therapy may occur and should be monitored for. Once controlled, the therapy is gradually reduced over 2–3 weeks. Intralesional corticosteroid injections will control persistent lesions. Systemic corticosteroids will result in rapid resolution of lesions, if not contraindicated by the underlying precipitating cause. In acute lesions, colchicine is often rapidly effective at a dose of 0.6 mg twice daily. In refractory cases, antimalarials such as hydroxycholoroquine may be tried. TNF-α medications may also be effective, but a paradoxic reaction in which adalimumab may have caused EN has been reported.
The prognosis in patients with acute EN is typically good, with the attack running its course in 3–6 weeks. Recurrences do occur, especially if the underlying condition or infection is still present, or if physical activity is resumed too quickly. Chronic or atypical lesions should suggest an alternative diagnosis and require a biopsy.
Acosta KA, et al: Etiology and therapeutic management of erythema nodosum during pregnancy. Am J Clin Dermatol 2013; 14: 215.
Blake T, et al: Erythema nodosum–a review of an uncommon panniculitis. Dermatol Online J 2014; 20.
Dengen A, et al: Erythema nodosum in a patient undergoing vemurafenib therapy for metastatic melanoma. Eur J Dermatol 2013; 23: 118.
Emre S, et al: A case of severe erythema nodosum induced by methimazole. Saudi Pharm J 2017; 25: 813.
Fruchter R, et al: Erythema nodosum in association with idiopathic granulomatous mastitis. J Eur Acad Dermatol Venereol 2017; 31: e391.
Ilhanli I, et al: Erythema nodosum during adalimumab therapy. Am J Internal Med 2015; 3: 210.
Kisacik B, et al: Multiclinical experiences in erythema nodosum. Rheumatol Int 2013; 33: 315.
Labunski S, et al: Tumour necrosis factor-alpha promoter polymorphism in erythema nodosum. Acta Derm Venereol 2001; 81: 18.
Mössner R, et al: Erythema nodosum-like lesions during BRAF inhibitor therapy. J Eur Acad Dermatol Venereol 2015; 29: 1797.
Passarini B, et al: Erythema nodosum. G Ital Dermatol Venereol 2013; 148: 413.
Uceda J et al: A6.15 refractory chronic erythema nodosum and treatment with anti TNF. Annals of the Rheumatic Diseases 2013; 72: A47.
Vargas-Hitos JA, et al: Erythema nodosum as azathioprine hypersensitivity reaction in a patient with bullous pemphigoid. Indian J Dermatol 2014; 58: 406.
Weizman A, et al: Clinical, serologic, and genetic factors associated with pyoderma gangrenosum and erythema nodosum in inflammatory bowel disease patients. Inflamm Bowel Dis 2014; 20: 525.
Lobular Panniculitis
Vessel-Based Lobular Panniculitis
Inflammation or thrombosis of blood vessels may lead to fat necrosis caused by ischemia. This can occur in primary forms of vasculitis, such as polyarteritis nodosa and Churg-Strauss syndrome, in metabolic disorders such as oxalosis and calciphylaxis, with atheromatous emboli, with heparin and coumarin necrosis, and with various coagulopathies. These entities are discussed in other chapters.
Nodular Vasculitis and Erythema Induratum
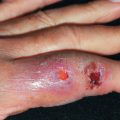
Clinically and histologically, nodular vasculitis is identical to erythema induratum (EI). The two differ only by the presence of TB as a precipitating factor in EI. Nodular vasculitis presents as tender, subcutaneous nodules on the calves of middle-aged women ( Fig. 23.3 ). Venous insufficiency may be present. Lesions are bilateral and less red and tender than EN; they often ulcerate, drain oily liquid, and recur over years.
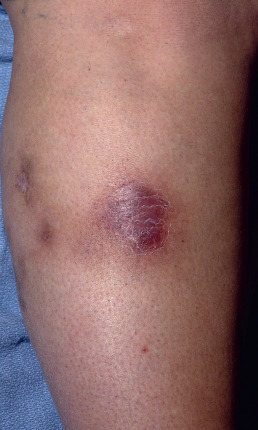
The early lesions may show a suppurative vasculopathy, proposed by various authors to be an arteritis, a venulitis, or both. In some cases, no vasculitis is found, and despite its name, the presence of a vasculitis is not required to establish the diagnosis. Nodular vasculitis results in substantial lobular necrosis of adipocytes with suppuration. Necrosis of the lobule results in loss of the lipocyte membrane and pooling of lipid into variably sized round aggregates. As lesions evolve, the fat becomes increasingly necrotic, forming microcysts, and the disease progresses to the point where it may perforate through the epidermis, forming ulceration. Granulomatous inflammation appears adjacent to areas of fat necrosis, and eventually lesions resolve with fibrosis.
Nontuberculous nodular vasculitis must be distinguished from EI. Nodular vasculitis has many reported causes, including Chlamydia infections, Crohn disease, Takayasu arteritis, BCG vaccination, TNF-α antagonist therapy, and non-TB mycobacterial infections. Because clinicopathologic features are identical in EI and nodular vasculitis, the differentiation is made by evaluating for tuberculous infection in the patient. A Quantiferon gold or tuberculin skin test can be administered. If this is positive, the appropriate diagnosis is EI. Polymerase chain reaction (PCR) of the affected tissue may reveal the DNA of M. tuberculosis in 50%–70% of cases of EI. As a tuberculid (inflammatory response to tuberculosis), EI is a manifestation of cellular immunity to TB, and the purified protein derivative (PPD) test will always be positive. PCR of the tissue is not recommended in patients who are tuberculin skin test negative. It should be noted that even in areas where TB is prevalent, EI is rare, representing only 1% of cutaneous manifestations of TB in one study. When present, EI may signal serious genitourinary involvement, including tuberculous epididymo-orchitis.
EI requires antibiotic therapy for the underlying TB. Treatment of nodular vasculitis is usually supersaturated solution of potassium iodide (SSKI) supersaturated solution of potassium iodide, as outlined for EN, and is effective in about half of patients. In the others, trials of colchicine, antimalarials, NSAIDs, mycophenolate mofetil, and systemic corticosteroids have been helpful. Support stockings, elevation, and treatment of associated venous insufficiency may also improve nodular vasculitis.
Chen S, et al: Mycobacterium tuberculosis infection is associated with the development of erythema nodosum and nodular vasculitis. PLoS One 2013; 8: e62653.
Gilchrist H, et al: Erythema nodosum and erythema induratum (nodular vasculitis). Dermatol Ther 2010; 23: 320.
Kabuto M, et al: Erythema induratum (nodular vasculitis) associated with Takayasu arteritis. Eur J Dermatol 2017; 27: 410.
Misago N, et al: Erythema induratum (nodular vasculitis) associated with Crohn’s disease. Am J Dermatopathol 2012; 34: 325.
Papathemeli D, et al: Explosive generalization of nodular vasculitis– Mycobacterium marinum challenges the paradigm. J Eur Acad Dermatol Venereol 2016; 30: e189.
Park SB, et al: Nodular vasculitis that developed during etanercept (Enbrel) treatment in a patient with psoriasis. Ann Dermatol 2015; 27: 605.
Sekiguchi A, et al: Erythema induratum of Bazin associated with bacillus Calmette–Guérin vaccination. J Dermatol 2016; 43: 111.
Taverna JA, et al: Case reports: nodular vasculitis responsive to mycophenolate mofetil. J Drugs Dermatol 2006; 5: 992.
Wee E, Kelly RI: Treatment of nodular vasculitis with colchicine. Australas J Dermatol 2017; 58: e79.
Lipodermatosclerosis
Lipodermatosclerosis, or sclerosing panniculitis, occurs primarily on the medial lower third of the lower legs of women older than 40 ( Fig. 23.4 ), with an above-average body mass index (BMI). It is often bilateral. In the acute phase, red to purple, poorly demarcated, indurated plaques are present on the lower legs. The lesions are painful and the differential diagnosis includes cellulitis, phlebitis, EN, or inflammatory morphea. In the chronic phase there is marked woody induration in a stocking distribution, resulting in calves that resemble inverted champagne bottles. This thick, tight, hyperpigmented skin results from fibrosis in the subcutaneous fat, which may occur without the primary inflammatory panniculitis ever being clinically observed. Fibrosis occurs multifocally and microscopically throughout the affected area.
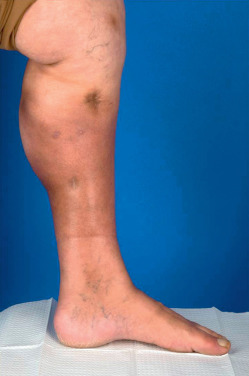
The etiology of lipodermatosclerosis is venous insufficiency. These patients may have venous varicosities, superficial thrombophlebitis, deep venous thrombosis, or several of these conditions. Even when venous disease is not clinically evident, evaluation of the venous system of the lower leg will frequently reveal insufficiency. Laboratory evaluation is rarely done but may reveal a genetic mutation in the fibrinolytic system resulting in increased thrombosis in these patients. Venous insufficiency results in hypoxia, necrosis of fat, inflammation, and eventual fibrosis. If hypoxemia is present from other causes, such as pulmonary disease, sclerosing panniculitis may be more severe. Angiosarcoma has been reported as a rare complication in the setting of postphlebitic lipodermatosclerosis.
The histologic features of sclerosing panniculitis are characteristic, but not all features may be seen on every biopsy, because the histologic features change over time within the lesion. The overlying dermis frequently shows changes of stasis with nodular proliferation of thick-walled vessels, hemosiderin deposition, fibrosis, and atrophy. In early lesions, there is ischemic necrosis in the center of the fat lobules manifested as “ghost cells”—pale cell walls with no nuclei along with thickened septae. Pseudomembranous changes in the dermis may also be a clue to early disease. There is a sparse lymphocytic infiltrate in the fat septa. As the lesions evolve, the septa are thickened and fibrosed, and there is a mixed inflammatory infiltrate of lymphocytes, plasma cells, and macrophages. Foamy histiocytes are present around the areas of fat necrosis. Fat microcysts are characteristic (but not diagnostic) and appear as small cysts with feathery eosinophilic remnants of adipocytes lining the cyst cavity and resembling frost on a window, so-called lipomembranous fat necrosis. In lesions later, these microcysts collapse and are replaced by fibrosis. Despite these characteristic features, biopsy should be avoided in these patients. Biopsies heal poorly and may lead to chronic leg ulcers. The diagnosis can usually be made clinically. Noninvasive techniques such as magnetic resonance imaging (MRI) have been used to avoid poorly healing wounds related to a biopsy. If a biopsy must be performed, it should be from the most proximal edge of involvement.
This diagnosis can be clinically confirmed if a careful vascular evaluation is performed. The location on the lower medial calf is unusual for EN. Most other panniculitides favor the posterior midcalf. The gradual progression from the ankles proximally is characteristic of sclerosing panniculitis and not other forms of lobular panniculitis.
The treatment of lipodermatosclerosis may be difficult. The fibrosis may be irreversible. Graded compression stockings with elevation and standard treatments for venous insufficiency are most effective in this condition. Application of pressure dressings, such as an Unna boot, can produce dramatic, if temporary, improvement. Greater compression—Unna boot with Coban and a foam buttress (bolster material to apply extra pressure to the red inflamed area) or the Profore boot—can be beneficial. Unfortunately, some patients cannot tolerate compression because of the pain of the lesions. Intralesional triamcinolone and ultrasound therapy have been used, but this is most effective when used in conjunction with compression. Pentoxiphylline, may be useful, especially in patients not responding to compression and elevation alone, or in patients who are initially intolerant of compression dressings. The addition of hydroxychloroquine to pentoxiphylline may provide additional improvement. Apparently, by enhancing the fibrinolytic capacity of affected patients, stanozolol, or oxandrolone, may benefit some patients. This is rarely required, however, if appropriate pressure dressings are applied and the patient is able to take full doses of pentoxiphylline. Stanozolol and oxandrolone may be virilizing for women and should be avoided in women of childbearing age. Stanozolol may induce hepatitis. Surgical treatment of varicosities and incompetent perforators may result in dramatic improvement in some patients.
Ayele A, et al: Pseudomembranous changes in the dermis. J Cutan Pathol 2017; 44: 1070.
Balasubramanyam S, et al: Venous treatment of lipodermatosclerosis to improve ambulatory function. Dermatolog Surg 2017 Aug 29; ePub ahead of print.
Chan CC, et al: Magnetic resonance imaging as a diagnostic tool for extensive lipodermatosclerosis. J Am Acad Dermatol 2008; 58: 525.
Choonhakarn C, Chaowattanapanit S: Lipodermatosclerosis. J Am Acad Dermatol 2012; 66: 1013.
Choonhakarn C, et al: Lipodermatosclerosis. Int J Dermatol 2016; 55: 303.
Damian DL, et al: Ultrasound therapy for lipodermatosclerosis. Arch Dermatol 2009; 145: 330.
Jeong, KH, et al: Refractory lipodermatosclerosis treated with intralesional platelet-rich plasma. J Am Acad Dermatol 2011; 65: e157.
Jowett AJ, et al: Angiosarcoma in an area of lipodermatosclerosis. Ann R Coll Surg 2008; 90: W15.
Segura S, et al: Lipomembranous fat necrosis of the subcutaneous tissue. Dermatol Clin 2008; 26: 509.
Vesić S, et al: Acute lipodermatosclerosis. Dermatol Online J 2008; 14: 1.
Walsh SN, et al: Lipodermatosclerosis. J Am Acad Dermatol 2010; 62: 1005.
Physical Panniculitis
The category of panniculitis includes processes in the fat that occur from physical factors. Some are characterized by the presence of needle-like clefts: sclerema neonatorum, subcutaneous fat necrosis, and poststeroid panniculitis. Infants and children are most frequently affected likely in part due the higher amount of brown fat in children. Fat is typically in a liquid form in lobules. Brown fat freezes into a solid at higher temperatures than regular fat in adults, so slight cooling can lead to solidification and necrosis.
Sclerema Neonatorum
Sclerema neonatorum is the most severe and rarest disorder of the physical panniculitides. It affects premature neonates who are critically ill for other reasons or have experienced profound hypothermia. With more adequate neonatal intensive care, this disorder has become extremely rare. Neonates affected with sclerema neonatorum usually die, unless the underlying diseases can be reversed. In the first few days of life, the skin begins to harden, usually initially on the buttocks or lower extremities, then rapidly spreads to involve the whole body. The skin on the palms, soles, and genitalia is spared. The skin becomes dry, livid, cold, rigid, and boardlike, limiting the mobility of the parts. The skin in the involved areas cannot be picked up. The skin of the entire body may appear half-frozen and is yellowish white. Visceral fat may also be involved. Therapy is mostly supportive, but some data suggest exchange transfusion may improve survival.
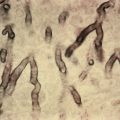
Histologically, adipocytes are enlarged and filled with needle-like clefts in a radial array Recently, this unusual histologic finding was documented in a reaction to gemcitabine. Affected fat cells undergo necrosis. There is sparse inflammation, and histiocytes containing needle-like clefts are rare, possibly because most children die before granulomas can form.
Subcutaneous Fat Necrosis of the Newborn
Subcutaneous fat necrosis of the newborn (SCFN) occurs during the first 4 weeks of life (half in the first week) in term or postterm infants. There is often a history of fetal distress such as birth asphyxia, maternal-fetal disproportion, or meconium aspiration. More recently, intensive care unit protocols for neonates with neural depression may initiate hypothermia, which can lead to widespread SCFN that can even involve the viscera. Septicemia, severe neonatal anemia, thrombocytopenia, and maternal cocaine use have also been associated with SCFN.
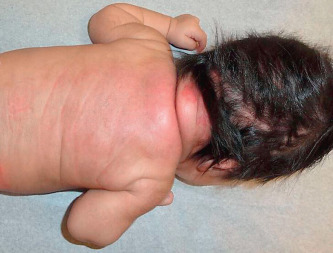
SCFN is characterized by painful, firm to rubbery, erythematous nodules usually on areas that have significant subcutaneous tissue and are susceptible to crush injury during birth such as the upper back, buttocks, cheeks, or proximal extremities ( Fig. 23.5 ). The arms, shoulders, and back were most common in a recent larger series. Lesions may fuse to form plaques. They heal spontaneously within 3 months with no scarring unless there is ulceration. In general, the infants remain well; however, hypoglycemia, thrombocytopenia, hypertriglyceridemia, lactic acidosis, and potentially life-threatening hypercalcemia may rarely occur. Some degree of hypercalcemia occurred in more than 50% of recently reported cases. The hypercalcemia may appear weeks to months after the appearance and resolution of the skin lesions. Periodic measuring of serum calcium for the first 3–6 months of life or until the lesions resolve has been recommended as the calcium can increase even as the lesions are resolving. Hypercalcemia may result in failure to thrive, irritability, lethargy, hypotonia, seizures, and renal failure. Rarely nephrocalcinosis and calcinosis of the gallbladder were reported. Significant hypercalcemia is treated with hyperhydration, calcium-wasting diuretics (furosemide), and formulas low in calcium and vitamin D. Systemic corticosteroids, calcitonin, and bisphosphonates may also be effective when other methods fail to reduce the hypercalcemia.