In this chapter proliferations derived from vascular endothelial cells, fibroblasts, myofibroblasts, smooth muscle cells, Schwann cells, and lipocytes are reviewed. Also discussed are several neoplasms of cells invading or aberrantly present in the dermis, such as metastatic cancer, endometriosis, and meningioma.
Cutaneous Vascular Anomalies
Vascular anomalies are overgrowths of various types of blood or lymphatic vessels. They can either occur in isolation or as part of a syndrome caused by a localized or generalized genetic mutation.
The International Society for the Study of Vascular Anomalies (ISSVA) created a classification system that has become the standard way of categorizing vascular lesions ( http://www.issva.org/UserFiles/file/Classifications-2014-Final.pdf ). Vascular lesions can be divided into vascular tumors that are dynamic and can exhibit rapid growth and malformations that are more static but can still grow. Recently antiproliferative medical therapy such as sirolimus has been found to be effective for some vascular anomalies, indicating that many of the malformations are actually more dynamic than originally thought.
Vascular Tumors
Benign Tumors
Infantile Hemangioma
Infantile hemangiomas (IHs) are the most common benign tumors of childhood, occurring in approximately 4%–5% of children. Importantly, IHs are either not detectable at birth or present with a patch of vasoconstriction called the “premonitory sign,” or a slightly bruised or telangiectatic. Many other vascular tumors such as kaposiform hemangioendotheliomas, congenital hemangiomas, fibrosarcomas, and rhabdomyosarcomas can be fully formed tumors at birth, and this can help differentiate them from IH. As the vessels of the IH form, a rim of vasoconstriction can often be seen at the edge even if it was not present from the start.
Risk factors for IH include low birth weight, female gender, multiplets (twin, triplets), prematurity, and maternal age over 30, although low birth weight seems to be the strongest predictor and the other factors may be confounded by the low birth weight associated with them. IH happen in all ethnicities and in all cutaneous locations. The majority of IHs occur sporadically and there is no known genetic predisposition for hemangiomas.
The pathogenesis of IHs is complex and not fully elucidated. CD133+ stem cells within the hemangioma differentiate into mature blood vessels that express GLUT-1, a glucose transporter normally restricted to endothelial cells with blood-tissue barrier function, as in brain and placenta. The vessels proliferate, then involute. Some suggest the stem cells could originate from placental trophoblast. Histologically, IHs are composed of primitive endothelial cells that are similar to placenta. Ultrastructurally, they lack typical Weibel-Palade bodies but do have crystalloid inclusions typical of embryonic endothelium and stain for GLUT-1. They also stain for Fc-γ-RII, Lewis Y antigen (LeY), and merosin. Early hemangiomas show evidence of endothelial progenitor cells that stain with CD133 and CD34. In late stages, the endothelium flattens, and the lumina are more apparent because of increased blood flow. In time, fibrosis becomes pronounced as involution progresses.
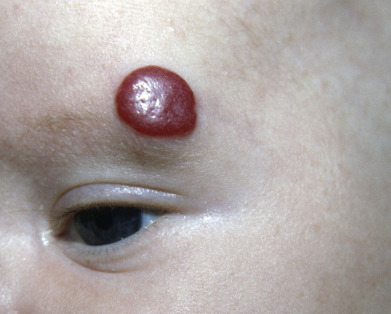
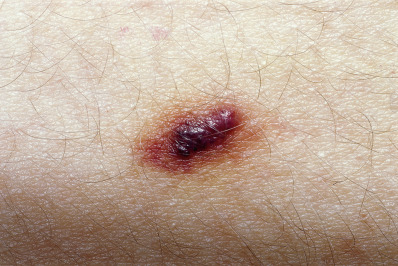
IHs can present anywhere on the body and can be divided based on multiple features. Superficial IHs are easily seen on top of the skin and often have a bright red or maroon look when proliferating ( Fig. 28.1 ). Deep hemangiomas are under the skin and often have a blue hue but are soft. Mixed hemangiomas have features of superficial and deep hemangiomas. Localized hemangiomas are round or oval and have well-defined margins. Segmental or patterned hemangiomas have less distinct borders and appear to grow in embryologic segments. Segmental hemangiomas can be associated with multiple syndromes as outlined later.
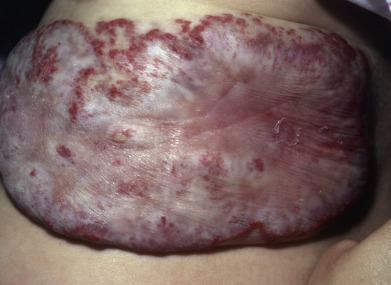
IHs go through a characteristic growth pattern with rapid growth occurring most commonly in the first 6 months, although they can grow up to 12 months. The most rapid growth of IH is between 5.5 and 7.5 weeks of life (no matter what the age of gestation) and 90% are finished significantly growing by 4 months, although larger lesions can continue to grow up to a year of life and very rarely longer. Therefore therapy, when needed, is ideally started early in life to prevent the rapid growth that can lead to tissue distortion. IHs then regress and although the majority of the regression happens by age 4 ( Fig. 28.2 ), continued improvement can occur up to and past age 10.
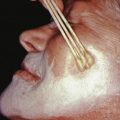
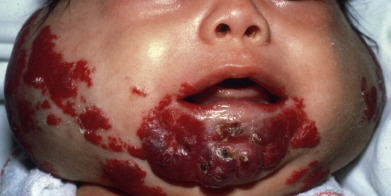
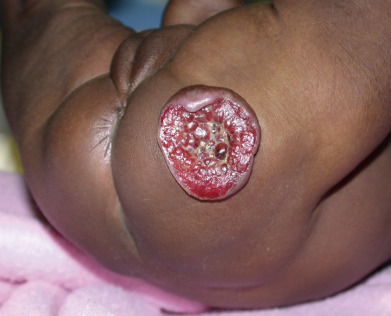
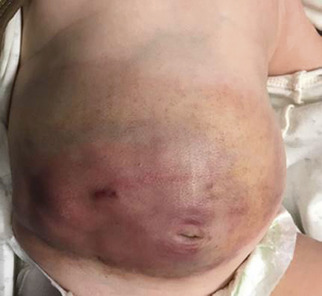
Complications of hemangiomas are related mostly to their location and typically due to physical distortion of the tissue or ulceration. Lesions around the eye can lead to amblyopia or astigmatism and should be co-managed with an ophthalmologist. Nasal tip lesions can lead to permanent distortion of the nasal tissue, collapse of the columella, or obstruction of the nasal passages, impairing breathing and almost always require early therapy to prevent this. Hemangiomas (especially segmental) in the jawline or neck (“beard” distribution) can be associated with airway involvement with IH ( Fig. 28.3 ). These patients present with inspiratory stridor in early infancy, often when there is severe airway occlusion or when there is a superimposed infection leading to airway inflammation. Airway IHs are often misdiagnosed as croup, especially because systemic steroids given for croup will shrink the hemangioma and thus ameliorate the symptoms. A lateral neck x-ray can show airway constriction, but direct visualization by an otorhinolaryngologist is necessary if an airway hemangioma is suspected. Perineal hemangiomas, lip hemangiomas, and those with a large or rapidly growing superficial component are at higher risk for ulceration ( Fig. 28.4 ). Segmental hemangiomas can ulcerate even if there is very little cutaneous component.
The decision to treat a hemangioma is in part based on how completely a hemangioma will regress without therapy. A hemangioma with a very sharp angle at the skin (similar to a mushroom cap) will leave more redundant tissue than a hemangioma with a gentle slope. Central forehead, central cheek, nasal tip, and lip IHs are more likely to leave permanent distortion due to the need for these areas to have more taught skin, and treatment is often necessary to prevent this. The skin may appear normal after involution, but larger and more pedunculated lesions are more likely to leave atrophy, telangiectasia, or anetoderma-type redundancy. If a hemangioma ulcerates, there is invariably a scar.
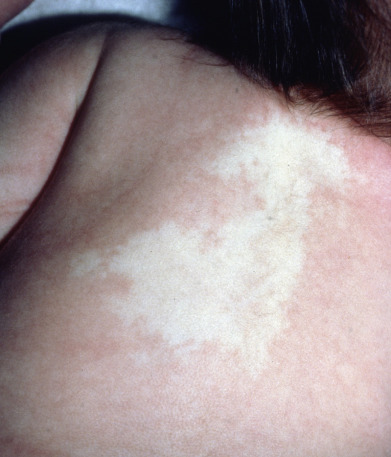
There is a variant of IH that is present at birth as a flat patch of telangiectasia or vasoconstriction but proliferates very little or not at all. These IHs have been named infantile hemangiomas with minimal or arrested growth (IH-MAG) or “abortive” hemangiomas. IH-MAG are manifested by a congenital patch of telangiectatic blood vessels and often the premonitory sign around them. They are GLUT-1 positive and may have small areas of typical hemangioma that show up within the original telangiectatic patch, and they tend to regress in the same time frame as IH but can also ulcerate. IH-MAG are important to recognize and differentiate from capillary malformations because they also have associated risk of hemangioma syndromes such as PHACE and LUMBAR syndrome (see later in this chapter) depending on location. Therapy for IH-MAG is often not necessary, but the same therapies that work for regular IHs will work for IH-MAG.
Simple observation may be appropriate for many hemangiomas, allowing the lesions to regress spontaneously. Parents should be informed of the appropriate time frame and the pros and cons should be explained. Indications for therapy include ulceration and threatened interference with vital functions (e.g., feeding, vision, respiration, passage of urine or stool). Specifically, there is a risk of occlusion amblyopia, astigmatism, and myopia from periorbital hemangiomas, as well as interference with limb function, tissue destruction, or cardiovascular compromise from high-output cardiac failure. Strong consideration should be given to treatment of hemangiomas that may lead to permanent disfigurement or long-term psychological consequences, such as large hemangiomas of the ear, nose, glabellar area, or lips.
For therapy, β-blockers are used most frequently, but systemic treatment can rarely be complicated by bradycardia or hypoglycemia, and regular feedings are critical before and during treatment. Propranolol is now approved by the U.S. Food and Drug Administration (FDA) for hemangiomas in children over 45 weeks of age (e.g., 40 weeks of normal gestation plus 5 weeks of infancy would equal 45 weeks). Atenolol and other more selective β-blockers have been used in patients who have concerns about propranolol in terms of its risk of bronchoconstriction and sleep disturbances. Propranolol crosses the blood-brain barrier, so some have argued that other β-blockers may have a lower risk of cognitive or behavioral impairment, although long-term studies do not seem to show any learning disabilities in children who have been given propranolol for hemangiomas. Side effects of propranolol include hypotension, bradycardia, and hypoglycemia. Symptoms of hypoglycemia may be masked by the propranolol because it blocks the tachycardia and sweating that often alerts someone to his or her hypoglycemia. It is extremely important that the child has been fed immediately before the propranolol administration to avoid hypoglycemia. If children are vomiting or having diarrhea or not feeding well, the medicine should be held. Protocols for starting propranolol typically involve checking a baseline heart rate and blood pressure and then starting oral propranolol at 1 mg/kg/day divided twice daily at least 10 hours apart (0.5 mg/kg/dose). If the vital signs are normal 1 and 2 hours after starting this dose, it is continued for a week and then the patient is uptitrated to 2 mg/kg/day divided twice daily. In the same manner and if clinically indicated this can be repeated a week later up to 3 mg/kg/day divided twice daily. Regrowth of treated hemangiomas is frequently seen and seems to be more prominent the sooner the propranolol is stopped. Most authors will treat children until around 12–15 months of age and if there is rebound, the medication can be restarted. There are rarely children who need to stay on propranolol for multiple years and even more rarely children whose hemangiomas regrow somewhat a year or more after stopping the medicine.
Some hemangiomas respond to topical β-blockers. Topical timolol, although off label, is extremely effective for superficial hemangiomas and can help heal ulcerations. The timolol gel-forming solution 0.5% is typically used, but caution is advised in premature infants and ulcerations. Timolol is approximately 10 times more potent per drop than propranolol and some advocate that similar precautions should be taken as for propranolol. Timolol is absorbed especially through the scalp and likely through mucous membranes or ulcerations. One drop is applied to the hemangioma 2 or 3 times per day.
Oral prednisolone at a dose of 2–4 mg/kg/day has also been used for IH but has mostly been replaced by propranolol. In the patients who respond well to treatment, the enlarging hemangioma stops growing in 3–21 days. The prednisolone is continued for 30–90 days. Systemic steroids are still used if propranolol is contraindicated or in conjunction with propranolol in hemangiomas that need to be treated emergently. The use of both medications concomitantly is extremely effective but when tapering the prednisolone, hypoglycemia has been reported. Intralesional corticosteroid treatment has been used but carries some risk of embolization and occlusion of ocular vessels. Injection regularly produces pressures exceeding the systemic arterial pressure, leading to possible embolization. Selective arterial embolization may be necessary for extremely large lesions such as in life-threatening situations.
Treatment with recombinant interferon is not used because of the risk of spastic diplegia. Topical imiquimod, low-frequency ultrasound, and ultrapotent topical steroids have been used historically before the realization of the efficacy of β-blockers. Both neodymium-doped yttrium-aluminum-garnet (Nd:YAG) and potassium titanyl phosphate (KTP) lasers have been used to deliver intralesional therapy.
There are two hemangioma syndromes that are also associated with large segmental hemangiomas of specific locations. Segmental hemangiomas on the head, neck, and proximal upper shoulder/neck areas, especially if over 5 cm in length, can be associated with PHACE syndrome. Larger hemangiomas seem to have higher risk. PHACE denotes the association of posterior fossa brain malformations (primarily the Dandy-Walker malformation), large segmental hemangiomas, arterial anomalies, coarctation of the aorta and other cardiac defects, and eye abnormalities. Most patients do not exhibit all of the features of PHACE. Midline defects of the chest and abdomen such as sternal clefting or complete sternal absence, as well as supraumbilical raphe, can be seen, so an S is sometimes added to the acronym: PHACE(S). The face can be divided into four embryologic segments: S1 is the forehead lateral to the medial canthus including the upper eyelid, S2 is the midcheek, S3 is the jawline, and S4 is a stripe down the central face from forehead to chin. A fifth segment has been recently proposed that involves the ocular orbit because an isolated hemangioma of the orbit can be associated with PHACE. PHACE workup involves evaluating for all of the possible anomalies, therefore the following should be done:
- 1.
An echocardiogram with special attention to the ascending aorta. Cardiac defects described include coarctation, aortic arch abnormalities, aberrant subclavian artery origin, vascular ring (less common), and interrupted or double aortic arch.
- 2.
Magnetic resonance imaging (MRI) of the brain for evaluation of the posterior fossa and including fine cuts of the orbits if there is concern for an orbital hemangioma: posterior fossa abnormalities (Dandy-Walker), hypoplasia of medulla or pons, absent pituitary, or intracranial hemangioma.
- 3.
Evaluation of the cervical and intracerebral blood vessels with MRI/magnetic resonance angiogram (MRA) of the head and neck: abnormal carotid, subclavian, or brachiocephalic arteries.
- 4.
Ophthalmologic evaluation: “morning glory” disc, abnormal or persistent fetal vasculature.
Further imaging of the chest may be needed if the hemangioma extends down, as well as consultation with appropriate subspecialties depending on the findings of the imaging. Evaluate for any associated endocrinologic abnormalities, including hypopituitarism, hypogonadism, hypothyroidism, or growth hormone deficiency. Headaches have been reported at higher levels. Patients should also be followed for normal neurodevelopment.
Propranolol use is debated in PHACE. Some patients with PHACE have had stroke after starting propranolol. Ideally imaging is done before start to rule out significant enough vascular changes that there may be a higher risk of stroke. However, most agree that if the hemangioma is high risk (vision threatening, airway, ulcerating), lower-dose propranolol can be started while starting the process of imaging.
LUMBAR syndrome (also called SACRAL and PELVIS) is the constellation of a large segmental perineal hemangioma with associated spinal dysraphism, renal anomalies, bony anomalies, and gastrointestinal (GI) anomalies. The acronym LUMBAR syndrome has been used to describe the association of lower body hemangioma, urogenital anomalies, myelopathy, bony deformities, anorectal malformations, arterial anomalies, and renal anomalies. In patients with a segmental hemangioma in the perineum, imaging with ultrasound or MRI should be considered to rule out abnormalities of the urogenital or renal systems. Segmental hemangiomas, especially if larger than 5 cm involving the area above the intergluteal cleft (often seen as a part of LUMBAR), are an extremely high-risk marker of spinal cord tethering. Ultrasound may not be sensitive to diagnose some abnormalities, and MRI is recommended in those patients so that any necessary repair can happen before permanent damage.
Children with five or more hemangiomas may have hemangiomas involving internal organs, most commonly the liver. Liver ultrasound is recommended in these patients. The individual lesions are smaller in size ranging from 1–10 mm and there can rarely be hundreds of lesions. Visceral lesions may be present in the central nervous system (CNS), lungs, liver, or other organs, leading to obstructive jaundice, internal bleeding, or respiratory failure. Having a few small lesions in the liver may not have any sequelae and can often be monitored to ensure resolution. More extensive involvement of the liver can lead to shunting of cardiac output leading to high-output failure. Therapy can be rapidly beneficial to improve cardiac function. Hemangiomas secrete type III deiodinase, which is an enzyme that converts active T3 and T4 to reverse T3. If there is a large enough volume of hemangiomas, an infant can become floridly hypothyroid requiring intravenous repletion. Single large hemangiomas in the liver are typically congenital hemangiomas (see later section).
Borok J, et al: Safety and efficacy of topical timolol treatment of infantile haemangioma. Br J Dermatol 2017 Aug 3; Epub ahead of print.
Brosig CL, et al: Neurodevelopmental outcomes in children with PHACE syndrome. Pediatr Dermatol 2016; 33: 415.
Chamlin SL, et al: Multicenter prospective study of ulcerated hemangiomas. J Pediatr 2007; 151: 684.
Garzon MC, et al: PHACE syndrome. J Pediatr 2016; 178: 24.
Hochman M: Infantile hemangiomas. Facial Plast Surg Clin North Am 2014; 22: 509.
Hoff SR, et al: Head and neck vascular lesions. Otolaryngol Clin North Am 2015; 48: 29.
Iacobas I, et al: LUMBAR. J Pediatr 2010; 157: 795.
Kanada KN: A prospective study of cutaneous findings in newborns in the United States. J Pediatr 2012; 161: 240.
McCuaig CC, et al: Therapy of ulcerated hemangiomas. J Cutan Med Surg 2013; 17: 233.
Munden et al: Monitoring propranolol treatment in periocular infantile haemangioma. Eye (Lond) 2014; 28: 1281.
Pope E, et al: Oral versus high-dose pulse corticosteroids for problematic infantile hemangiomas. Pediatrics 2007; 119: e1239.
Püttgen K, et al: Topical timolol maleate treatment of infantile hemangiomas. Pediatrics 2016; 138: e20160355.
Shehata N, et al: Late rebound of infantile hemangioma after cessation of oral propranolol. Pediatr Dermatol 2013; 30: 587.
Yu J, et al: Prevalence and clinical characteristics of headaches in PHACE syndrome. J Child Neurol 2016; 31: 468.
Rapidly Involuting Congenital Hemangioma and Noninvoluting Congenital Hemangioma
Rapidly involuting congenital hemangioma (RICH) and noninvoluting congenital hemangioma (NICH) are rare GLUT-1–negative vascular tumors that present fully grown at birth and either involute rapidly or fail to involute. Activating mutations in GNA11 and GNAQ have been demonstrated in these congenital lesions. Because many lesions only partially involute, some are deemed partially involuting congenital hemangioma (PICH). Children with RICH or NICH and IH have been described, but because IH is so common this is likely coincidence. Clinically most NICH lesions are flatter plaques and most RICH lesions are more raised and nodular. Systemic steroids and propranolol have been tried without success. If therapy is necessary for an ulcerated or bleeding lesion surgery or embolization has been used successfully.
A new type of congenital hemangioma with fetal involution has been proposed. This may be a RICH that involutes partially or entirely before birth. These lesions leave behind atrophy once resolved.
Ayturk UM, et al: Somatic activating mutations in GNAQ and GNA11 are associated with congenital hemangioma. Am J Hum Genet 2016; 98: 789.
Lu H, et al: A rare atypical rapidly involuting congenital hemangioma combined with vascular malformation in the upper limb. World J Surg Oncol 2016; 14: 229.
Maguiness S, et al: Rapidly involuting congenital hemangioma with fetal involution. Pediatr Dermatol 2015; 32: 321.
Sur A, et al: Multiple successful angioembolizations for refractory cardiac failure in a preterm with rapidly involuting congenital hemangioma. AJP Rep 2016; 6: e99.
Pyogenic Granuloma
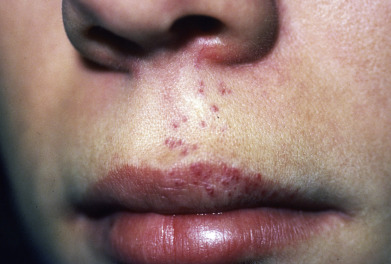
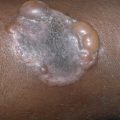
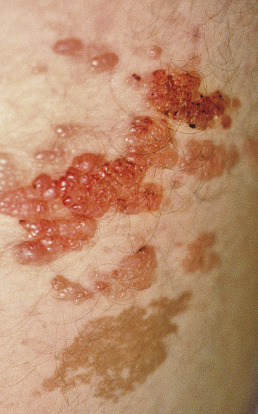
A pyogenic granuloma is a small, eruptive, usually solitary, sessile or pedunculated, friable papule ( Fig. 28.5 ). The lesion is common in children but may occur at any age, especially during pregnancy or with medication use as described later. Pyogenic granuloma occurs most often on an exposed surface: on the hands, forearms, or face, or at sites of trauma. The lesions can also occur in the mouth, especially on the gingiva, most often in pregnant women (granuloma gravidarum). On the sole of the foot or nail bed, it may be mistaken for a melanoma. Pyogenic granulomas bleed easily on the slightest trauma and, if cut off superficially, promptly recur. Recurring lesions may have one or many satellite lesions.
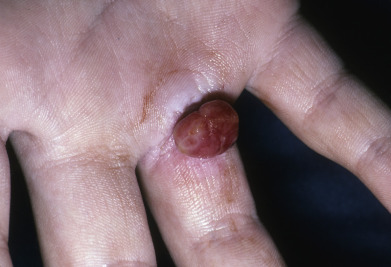
Pyogenic granulomas may be seen in patients treated with isotretinoin, capecitabine, vemurafenib, or indinavir. Isotretinoin treatment of acne vulgaris can be complicated by numerous exuberant pyogenic granuloma–like lesions of the trunk or periungual lesions. Some data suggest that patients with pyogenic granuloma have a statistically higher prevalence of Bartonella seropositivity compared with controls, but a definite etiologic role has not been established.
Histologically, pyogenic granuloma is a lobular capillary hemangioma, with lobules separated by connective tissue septa. With time, the epidermis becomes thinned, then eroded. Heavy secondary staphylococcal colonization is common. Intravascular pyogenic granuloma appears as a lobular capillary proliferation within a vein.
Treatment is by curettage or shave excision, followed by destruction of the base by fulguration or aluminum chloride. Silver nitrate alone may be sufficient to treat smaller lesions but may leave a silver tattoo, and it does not allow histopathologic evaluation to rule out a bleeding amelanotic lesion such as a melanoma. Topical timolol, imiquimod under occlusion, and sclerotherapy with monoethanolamine oleate or sodium tetradecyl sulfate have been used successfully. At times, a recalcitrant lesion may require excision or laser ablation. The drug-induced variety will regress after lowering of the dose or discontinuation of the medication. Systemic corticosteroids have been used to treat recurrent giant pyogenic granulomas.
Samatha Y, et al: Management of oral pyogenic granuloma with sodium tetradecyl sulphate. NY State Dent J 2013; 79: 55.
Sammut SJ, et al: Pyogenic granuloma as a cutaneous adverse effect of vemurafenib. N Engl J Med 2014; 371: 1265.
Locally Aggressive or Borderline Vascular Tumors
These tumors can proliferate more aggressively and show borderline malignant potential.
Hemangioendotheliomas
Hemangioendotheliomas (HEs) are a group of tumors that span the spectrum from benign to low-grade malignancy.
Kaposiform Hemangioendothelioma
Kaposiform hemangioendothelioma (KHE) is an uncommon vascular tumor that affects infants and young children. Rare cases have been reported in adults. It was first designated KHE in 1993. Although it frequently occurs in the retroperitoneum, KHE may present as multinodular soft tissue masses, purpuric macules, plaques, and multiple telangiectatic papules ( Fig. 28.6 ). The lesions extend locally and usually involve the skin, soft tissues, and even bone. The cutaneous variant may be associated with lymphangiomatosis. KHE is locally aggressive and may be complicated by platelet trapping and consumptive coagulopathy (Kasabach-Merritt syndrome), but distant metastases have not yet been reported. It has also been reported in association with Milroy-Nonne disease (primary hereditary lymphedema).
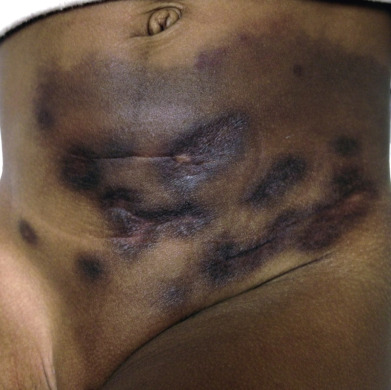
Histologically, there are combined features of cellular IH and Kaposi sarcoma. Additionally, in some tumors, lymphangiomatosis is seen sharply separated from the vascular lesion. There is a multilobular appearance that closely resembles that of tufted angioma, but in KHE, lesions are larger and less circumscribed and involve the deep soft tissue and even bone. Transition between these tumors has been described.
The prognosis depends on the depth and location of the lesion. Significant morbidity and mortality may result from compression and invasion of surrounding structures. If localized to the skin, lesions may be successfully excised. However, because of their tendency for deep and infiltrative growth, this is usually not possible. Combination of systemic corticosteroids and vincristine is often used but is being replaced by rapamycin (sirolimus).
Kasabach-Merritt Syndrome
In Kasabach-Merritt syndrome (KMS) there is platelet trapping within the vascular tumor and this leads to activation of the clotting cascade and disseminated intravascular coagulation (DIC). Low-grade KMS can be happening subtly but can worsen rapidly with acute, extreme swelling and purple discoloration to the lesion and this can be life threatening. The fibrinogen and platelet counts are low, the activated partial thromboplastin time and prothrombin time will be prolonged, and the d -dimer is elevated, indicating DIC. Before the onset of the acute event, the infant will often have a reddish or bluish plaque or tumor on the limb or trunk or, in rare instances, no visible lesion at all. The lesions usually have an associated lymphatic component, and most are KHEs. KMS also occurs in tufted angiomas and multifocal lymphangioendotheliomatosis, lesions that both demonstrate lymphatic differentiation. Venolymphatic and lymphatic malformations can also cause platelet trapping with resultant DIC. KMS is rarely reported with angiosarcoma. Some patients with VMs will have a chronic low-grade consumptive coagulopathy that occurs throughout life, and RICH lesions can trap platelets but not induce KMS; these should not be confused with KMS. The original reports of KMS occurring in IHs are incorrect.
Infants with KMS suddenly develop a painful violaceous mass in association with purpura and thrombocytopenia ( Fig. 28.7 ). The most striking sign is the bleeding tendency, especially in the hemangioma itself or into the chest or abdominal cavities. The spleen may be enlarged. Hemoglobin, platelets, fibrinogen, and factors II, V, and VIII are all reduced. Prothrombin time and partial thromboplastin time are prolonged, and fibrin split products may be elevated. Cases of microangiopathic hemolytic anemia have also been described. Repeated episodes of bleeding may occur, and although these may be spontaneous, bleeding can be precipitated by surgery, directed either at the hemangioma or elsewhere. The mortality rate may be as high as 30%, with most deaths secondary to bleeding complications.
KMS may be a self-limited disorder but often requires therapy. Systemic corticosteroids seem to work quickly, but often maintenance therapy is needed to prevent recurrence. Sirolimus has also become a favored therapy due to its efficacy and rapid benefit. Low-dose aspirin (5–10 mg/kg/day) can be used to decrease the platelet trapping and seems to help prevent KMS in some patients. This is a reasonable maintenance plan, but in children, care must be taken to avoid concomitant vaccination with live vaccines (especially influenza and varicella) and stopping aspirin if either primary infection is suspected to avoid Reye syndrome. Some have argued that the dose of aspirin is lower than the threshold to cause Reye syndrome. Adding ticlopidine may provide more benefit.
Historically, interferon (IFN) alfa-2a, vincristine, vinblastine, cyclophosphamide, actinomycin D, embolization, ε-aminocaproic acid, antiplatelet agents, irradiation, excision, and compression therapy have been used, alone or in combination.
Drolet BA, et al: Consensus-derived practice standards plan for complicated kaposiform hemangioendothelioma. J Pediatr 2013; 163: 285.
Margolin JF, et al: Medical therapy for pediatric vascular anomalies. Semin Plast Surg 2014; 28: 79.
O’Rafferty C, et al: Recent advances in the pathobiology and management of Kasabach-Merritt phenomenon. Br J Hameatol 2015; 171: 38.
Spindle Cell Hemangioma (Spindle Cell Hemangioendothelioma)
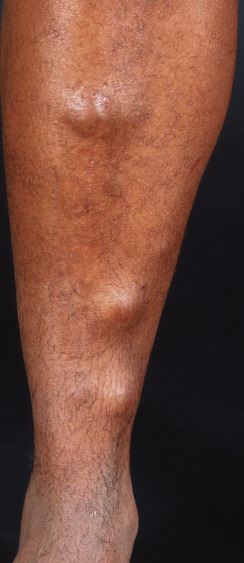
Spindle cell hemangioma is a vascular tumor that typically presents in a child or young adult with blue nodules of firm consistency on a distal extremity ( Fig. 28.8 ). Usually, multifocal lesions occur within an anatomic region. Histologically, a well-circumscribed dermal nodule will contain dilated vascular spaces with fascicles of spindle cells between them. Areas of the tumor will have an open alveolar pattern resembling hemorrhagic lung tissue. Phleboliths are common. A thrombosed, large, adjacent vessel with recanalization may be identified. The lesions appear to represent benign vascular proliferations in response to trauma to a larger vessel. They may recur after excision.
Epithelioid Hemangioendothelioma
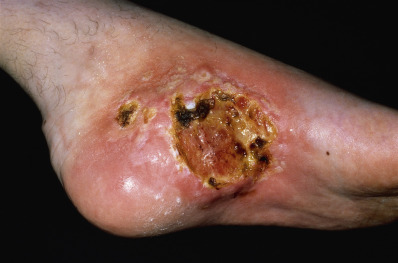
Epithelioid hemangioendothelioma usually presents as a solitary, slow-growing papule or nodule on a distal area of an extremity and behaves as a low-grade malignancy ( Fig. 28.9 ). There is a male preponderance, and onset is frequently before the individual is 25 years of age. Histologically, there are two components: dilated vascular channels and solid epithelioid and spindle-cell elements with intracytoplasmic lumina. It is caused by a fusion of WWTR1-CAMTA1; therefore CAMTA1 nuclear expression can differentiate it from other tumors. Wide excision is recommended with evaluation of regional lymph nodes, which are the usual site of metastases. In the minority of cases in which distant metastatic lesions develop, chemotherapy, radiation, or both may be employed.
Retiform Hemangioendothelioma
Retiform hemangioendothelioma is another form of low-grade malignancy that presents as a slow-growing exophytic mass, dermal plaque, or subcutaneous nodule on the upper or lower extremities of young adults. Histologically, there are arborizing blood vessels reminiscent of normal rete testis architecture. Human herpesvirus 8 (HHV-8) deoxyribonucleic acid (DNA) sequences have been reported in this tumor. Wide excision is recommended, although local recurrences are common. To date, no widespread metastases have occurred, although regional lymph nodes may develop tumor infiltrates.
Epithelioid Sarcoma–Like (Pseudomyogenic) Hemangioendothelioma
The epithelioid sarcoma–like variant demonstrates sheets of spindle, epithelioid, and rhabdomyoblastic cells. They can occur on the palms or soles and mimic a recalcitrant wart. This variant also behaves as a low-grade malignancy.
Endovascular Papillary Angioendothelioma (Dabska Tumor)
Endovascular papillary angioendothelioma, a rare low-grade angiosarcoma, presents as a slow-growing tumor on the head, neck, or extremity of infants or young children. It shows multiple vascular channels with papillary plugs of endothelial cells surrounding central, hyalinized cores that project into the lumina, sometimes forming a glomeruloid pattern. The entity is controversial; similar histologic features have been observed in other vascular tumors, such as angiosarcoma, retiform hemangioendothelioma, and glomeruloid hemangioma. The tumor may be a distinct entity or may demonstrate a histologic pattern seen in other vascular tumors. Wide excision and excision of the regional lymph nodes, when involved, are usually curative.
Doyle LA, et al: Nuclear expression of CAMTA1 distinguishes epithelioid hemangioendothelioma from histologic mimics. Am J Surg Pathol 2016; 40: 94.
Flucke U, et al: Epithelioid hemangioendothelioma. Diagn Pathol 2014; 9: 131.
Liau JY, et al: Composite hemangioendothelioma presenting as a scalp nodule with alopecia. J Am Acad Dermatol 2013; 69: e98.
McNab PM, et al: Composite hemangioendothelioma and its classification as a low-grade malignancy. Am J Dermatopathol 2013; 35: 517.
Requena L, et al: Cutaneous epithelioid sarcomalike (pseudomyogenic) hemangioendothelioma. JAMA Dermatol 2013; 149: 459.
Tufted Angioma (Angioblastoma)
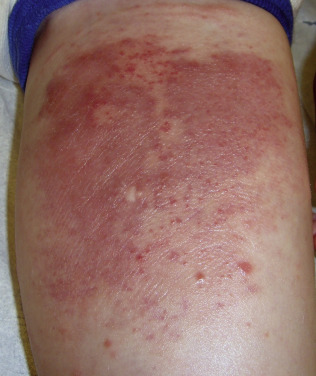
The tufted angioma lesion usually develops in infancy or early childhood on the neck and upper trunk. Adult onset has also been described. The lesions present as poorly defined, dull-red plaques with a mottled appearance, varying from 2–5 cm in diameter. Some show clusters of smaller angiomatous papules superimposed on the main macular area ( Fig. 28.10 ), and associated hypertrichosis has been noted. The lesions are usually sporadic, although familial cases have been reported. Histologic examination reveals small, circumscribed angiomatous tufts and lobules scattered in the dermis in a so-called cannonball pattern. Tumors with features of both tufted angioma and KHE have been described, and transformation between the tumors has also been noted. Immunostaining can be helpful in distinguishing these tumors. Tufted angioma is characterized by a proliferation of CD34+ endothelial cells with few actin-positive cells. KHE shows CD34 staining only in the luminal endothelial cells. In IH, GLUT-1 is positive.
Most lesions slowly extend with time, being progressive but benign in nature. Occasional spontaneous regression is documented. Therapy is dependent on the activity of the lesion. For small flat lesions aspirin may be enough but should be used with caution at low doses and stopped around the time of live vaccines such as varicella in young children to avoid Reye syndrome. For larger, growing lesions, especially with Kasabach-Merritt phenomenon, systemic steroids are rapidly effective. Vincristine is classically used, but sirolimus has proven to be very effective. Aspirin may then be used to maintain benefit more long-term. Treatment with pulsed dye laser, intense pulsed light, excision, and radiation has been successful. Lesions associated with KMS have also been treated with embolization, prednisone, and vincristine.
The term angioblastoma has also been used for a rare pediatric tumor often associated with destruction of regional structures, including bone. Basic fibroblast growth factor has been reported to be elevated, and some patients have responded to treatment with IFN alfa-2b.
Adams DM, et al: Efficacy and safety of sirolimus in the treatment of complicated vascular anomalies. Pediatrics 2016; 137: e20153257.
Fahrtash F, et al: Successful treatment of kaposiform hemangioendothelioma and tufted angioma with vincristine. J Pediatr Hematol Oncol 2010; 32: 506.
Javvaji S, et al: Response of tufted angiomas to low-dose aspirin. Pediatr Dermatol 2013; 30: 124.
Sabharwal A, et al: Acquired tufted angioma of upper lip. Head Neck Pathol 2013; 7: 291.
Wang L, et al: Congenital disseminated tufted angioma. J Cutan Pathol 2013; 40: 405.
Yamamoto Y, et al: Successful treatment of tufted angioma with propranolol. J Dermatol 2014; 41: 1120.
Hemangiopericytoma
True hemangiopericytomas are rare. The term is now reserved for lesions that demonstrate differentiation toward pericytes and cannot be otherwise classified. Most lesions formerly classified as hemangiopericytomas are now classified as examples of solitary fibrous tumor or giant cell angiofibroma. Remaining lesions can often be classified as glomangiopericytoma/myopericytoma or infantile myofibromatosis.
Clinically, hemangiopericytomas are nontender, bluish red tumors that occur on the skin or in the subcutaneous tissues on any part of the body. The firm, usually solitary nodule may be up to 10 cm in diameter. Histologically, the tumor is composed of endothelium-lined vessels that are filled with blood and surrounded by cells with oval or spindle-shaped nuclei (pericytes). The pericytes often form a concentric perivascular pattern. Staghorn-like ectatic spaces are often encountered. Wide local excision is the treatment of choice, but radiation therapy may produce excellent palliation.
Lesions Formerly Classified as Hemangiopericytomas
Various soft tissue tumors can present with a hemangiopericytoma-like staghorn vascular pattern, the most common being solitary fibrous tumor. Solitary fibrous tumor is usually CD34+ and has a wide distribution in the skin, mucosa, and viscera. When excision cannot be accomplished, targeted therapy, including imatinib, may be helpful. Myofibromas demonstrate nodular, pale-blue, hypocellular zones with surrounding hypercellular zones that contain staghorn vessels. Some examples lack the hypocellular zones and present only with a hemangiopericytoma-like pattern. Myopericytoma is a rare mesenchymal neoplasm that typically involves the extremities. The tumor demonstrates concentric perivascular spindle cells with myoid differentiation. Glomangiopericytoma is a closely related tumor composed of perivascular spindle cells with myoid differentiation; it combines features of glomus tumors and a hemangiopericytoma-like vascular pattern.
Stacchiotti S, et al: Targeted therapies in rare sarcomas. Hematol Oncol Clin North Am 2013; 27: 1049.
Watanabe K, et al: CD34-negative solitary fibrous tumour resistant to imatinib. BMJ Case Rep 2013 Jul 5; 2013.
Other Vascular Tumors
Cherry Angiomas (Senile Angiomas, de Morgan Spots)
These round, slightly elevated, ruby-red papules 0.5–6 mm in diameter are the most common vascular anomalies. It is a rare 30-year-old person who does not have a few, and the number increases with age. Probably every 70-year-old person has some senile angiomas. Most are on the trunk; they are rarely seen on the hands, feet, or face. Early lesions may mimic petechiae. When lesions are surrounded by a purpuric halo, amyloidosis should be suspected. Eruptive lesions have been described after nitrogen mustard therapy. Light electrodesiccation or laser ablation with intense pulsed light and long-pulse Nd:YAG laser systems can be effective. Shave excision can also be performed, but most patients accept reassurance and do not request removal.
Fodor L, et al: A side-by-side prospective study of intense pulsed light and Nd:YAG laser treatment for vascular lesions. Ann Plast Surg 2006; 56: 164.
Ma HJ, et al: Eruptive cherry angiomas associated with vitiligo. J Dermatol 2006; 33: 877.
Targetoid Hemosiderotic Hemangioma
In 1988 Santa Cruz and Aronberg described a lesion characterized by a central brown or violaceous papule surrounded by an ecchymotic halo ( Fig. 28.11 ). The term hobnail hemangioma has been proposed because many lesions are not targetoid. These acquired hemangiomas occur in young to middle-aged individuals and are present on the trunk or extremities. Because of their dark purple/black color they clinically simulate melanoma. The color likely represents trauma to a preexisting vascular lesion, with thrombosis and subsequent recanalization. Histologically, a biphasic growth pattern is seen, with central, superficial, dilated vascular structures lined by prominent hobnail endothelial cells, and collagen-dissecting, narrow vessels in deeper parts of the lesion. The endothelial cells commonly stain for CD31, but not CD34. D2-40 staining suggests lymphangiomatous proliferation.
Gutte RM, Joshi A: Targetoid hemosiderotic hemangioma. Indian Dermatol Online J 2014; 5: 559.
Glomeruloid Hemangioma
Glomeruloid hemangioma is a distinctive benign vascular neoplasm first described in 1990 and reported in patients with POEMS (Crow-Fukase) syndrome and Castleman disease. Some have also been associated with idiopathic thrombocytopenic purpura and Sjögren syndrome. Similar lesions have been reported in patients who are otherwise healthy.
The POEMS syndrome consists of polyneuropathy (severe sensorimotor), organomegaly (heart, spleen, kidneys), endocrinopathy, M component (M protein, monoclonal gammopathy), and skin changes (hyperpigmentation, hypertrichosis, thickening, sweating, clubbed nails, leukonychia, angiomas). Small, firm, red to violaceous papules appear on the trunk and proximal extremities in approximately one third of patients. Histologically, the lesions may be microvenular hemangiomas, cherry angiomas, multinucleated cell angiohistiocytomas, or glomeruloid hemangiomas. The latter consist of ectatic vascular structures containing aggregates of capillary loops within a dilated lumen, simulating the appearance of a renal glomerulus. Sequestered degenerating red blood cells are a characteristic finding. Two types of endothelial cell have been noted within the lesions: a capillary-type endothelium with large vesicular nuclei, open chromatin pattern, and a large amount of cytoplasm; and sinusoidal endothelium with small basal nuclei, dense chromatin, and scant cytoplasm. Lesions associated with POEMS syndrome demonstrate increased expression of vascular endothelial growth factor (VEGF) and its receptor, Flt-1.
Jacobson-Dunlop E, et al: Glomeruloid hemangiomas in the absence of POEMS syndrome. J Cutan Pathol 2012; 39: 402.
Eccrine Angiomatous Hamartoma
Eccrine angiomatous hamartoma usually appears as a solitary nodular lesion on the acral areas of the extremities, particularly the palms and soles, but identical lesions also occur on areas of the body that normally have few eccrine glands. This lesion appears at birth or in early childhood and is often associated with pain and hyperhidrosis. The lesion is a dome-shaped, tender, bluish nodule. Hypertrichosis may be present. When it is stroked or pinched, drops or beaded rings of perspiration may be seen.
Histologically, there is a combination of lobules of mature eccrine glands and ducts with thin-walled blood vessels. Excessive mucin, fat, smooth muscle, nerve infiltration, and terminal hairs may be present. The lesion has been associated with spindle cell hemangioma, arteriovenous malformation (AVM), and verrucous hemangioma. Excision may be necessary because of pain.
Halder C, et al: Eccrine angiomatous hamartoma. Indian J Dermatol 2014; 59: 403.
Shin J, et al: Eccrine angiomatous hamartoma. Ann Dermatol 2013; 25: 208.
Vascular Malformations
Malformations are abnormal structures that result from an aberration in embryonic development or trauma. The abnormality may be caused by an anatomic malformation or a functional alteration (as in nevus anemicus). Anatomic malformations are subdivided according to the type of vessel involved: capillary, venous, arterial, lymphatic, or combined. The term capillary malformation is best used as a term encompassing a variety of entities, including salmon patch, certain telangiectasias, port wine stains (PWSs), and cutis marmorata telangiectatica congenita. The characteristics of capillary malformations help differentiate various syndromes.
Happle R: What is a capillary malformation? J Am Acad Dermatol 2008; 59: 1077.
Lee MS, et al: Diffuse capillary malformation with overgrowth. J Am Acad Dermatol 2013; 69: 589.
Capillary Malformations
There are multiple different forms of capillary malformations (CMs), and each type can be seen in isolation or in association with a syndrome. CMs notably develop eczematous changes more easily over the top and will become darker red or purple with Valsalva maneuver or temperature changes. Differentiating the type of CM can help with diagnosis of the specific syndrome. Each of the different types of CM will be described and then the vascular syndromes that can present with CM will be discussed. Histologically, PWSs are made of dilated capillaries in the subpapillary network. Laser therapy has been used with satisfactory results, but a number of treatments are required, and recurrence years later usually requires some retreatment. The flashlamp pulsed dye 585- or 595-nm laser has the best record of safety and efficacy. Pulse durations are adjusted based on the size of the blood vessel and response to therapy. For darker-skinned patients, multiple pulse stacking with multiple cryogen spurts provides better epidermal protection. Long-pulse pulsed alexandrite lasers work best for hypertrophic, purple lesions, whereas pulsed dye lasers work best for flat, pink lesions. A frequency-doubled (532-nm) Nd:YAG laser that allows for shorter pulse widths, large spot sizes, and high fluences resulted in up to 75% improvement in color at 1 month after a single treatment. Topical sirolimus added to standard pulsed dye laser may augment therapy. The CMs associated with capillary malformation–arteriovenous malformation (CM-AVM) may have arterial flow and therefore laser may exacerbate them.
Nevus Simplex
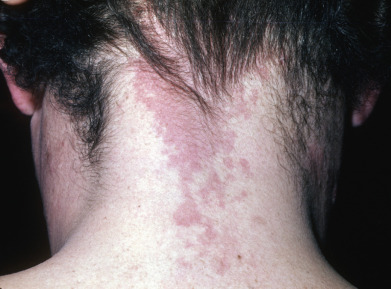
Nevus simplex is a type of CM that is light pink, is easily blanchable, often has a splattered appearance, is located in characteristic locations along the neural axis, and tends to fade over time. There have been many names used historically, including nevus flammeus and PWSs, but the nomenclature has been clarified by Frieden et al. recently. Nevus simplex malformations are common birthmarks in children, occurring in approximately 80% of children. They may persist in at least 5% of the population. Colloquial terms are used for certain types: “stork bite” is a pink-red macule situated on the posterior midline between the occipital protuberance and the tip of the spine of the fifth cervical vertebra ( Fig. 28.12 ); “angel’s kiss” is located on the glabella. Other typical locations are upper eyelid (15% of babies), along the nasal philtrum and ala, the temples, vertex scalp, upper back, and along the lumbar spine. Lesions in the lumbar area in isolation are typically not associated with spinal dysraphism but in association with other features can be a marker and necessitate imaging. Frieden proposed the term nevus simplex complex when children have very widespread lesions. Nevus simplex malformations tend to fade over time and are only rarely associated with syndromes (reviewed later in chapter): Beckwith-Wiedemann, Roberts SC, Nova, odontodysplasia, or megalencephaly CM syndrome.
Port Wine Stains
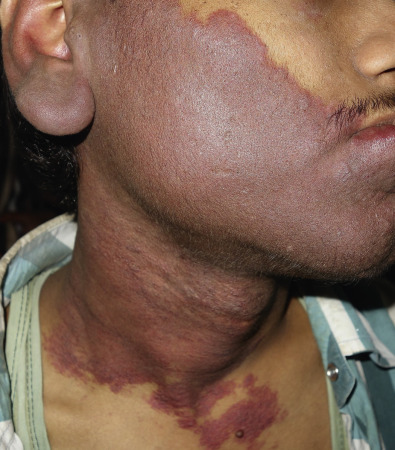
Port wine stains (PWSs) occur in an estimated 3 per 1000 children. The stains are present at birth and vary in color from pink to dark or bluish red. The lesions are usually unilateral, and although the face and neck are common locations ( Fig. 28.13 ), they can be located anywhere on the skin and may be widespread and involve as much as half the body. With facial lesions, the mucous membrane of the mouth may be involved. Although the surface of a PWS is usually smooth, small vascular nodular outgrowths or warty excrescences may develop over decades thus justifying early preventive intervention. These lesions often become more bluish or purple with age.
Rarely, PWS may appear as an acquired condition, usually with onset after trauma. The early inflammatory stage of morphea can also start with a purple patch that is mistaken for a PWS.
Reticulated Port Wine Stains
Reticulated PWSs are faint red-pink patches that have indistinct borders but often accentuate dramatically with Valsalva maneuver or temperature changes. They tend to be widespread and can be mistaken for cutis marmorata telangiectatica congenita (CMTC) (both lesions can occur in the same patient). Therefore some of the previous association between CMTC and systemic findings are likely mislabeled. Reticulated PWSs have been associated with various syndromes, including diffuse CM with overgrowth and some patients with PiK3Ca-related overgrowth syndromes (PROS), especially those with the megalencephaly capillary malformation (MCAP) phenotype.
Geometric Port Wine Stain
PWS can sometimes be dark purple and geometric with a sharp distinct cutoff. This CM phenotype is more associated with underlying venous or venolymphatic malformations such as can be seen in the following syndromes: Proteus syndrome; Klipell-Trenaunay syndrome; congenital lipomatous overgrowth with vascular anomalies, epidermal nevi, and skeletal, scoliosis, and spinal abnormalities (CLOVES) syndrome; and CLAPO syndrome. The geometric CM can have vascular and keratotic vascular papules within it.
Cutis Marmorata Telangiectatica Congenita
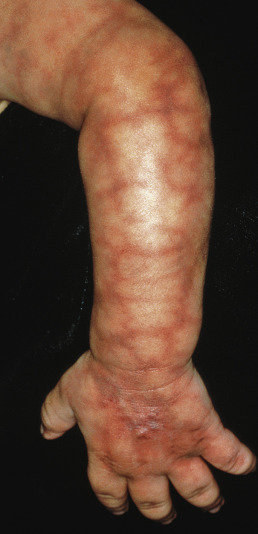
Cutis marmorata telangiectatica congenita (CMTC) has been recategorized as a type of capillary malformation. CMTC is characterized by the presence of a red-purple, livedo-appearing, reticulated, vascular network with a segmental distribution, usually involving the extremities ( Fig. 28.14 ). The mottling is pronounced and is made more distinct by crying, vigorous activity, and cold. There is often associated atrophy giving a “pseudoathletic” look that outlines the muscles. CMTC can be difficult to differentiate from a reticulated PWS, but typically CMTC has more uniformity and is more netlike. Lesions tend toward improvement with time but may not fully resolve. If located around the eye, some have advocated for ophthalmologic evaluation. The condition occurs sporadically, and there is a female preponderance. CMTC can be associated with phakomatosis pigmentovascularis and the Adams-Oliver syndrome (limb abnormalities, scalp defects, skull ossification defects).
Syndromes Associated With Capillary Malformations
Sturge-Weber Syndrome
PWS on the upper face may be a marker for Sturge-Weber syndrome (SWS; encephalotrigeminal angiomatosis) ( Fig. 28.15 ). There is debate as to what pattern these PWSs are following and what locations portend the highest risk. Originally PWSs were described as following nerve distributions (V1, V2, etc.), but more recently it has been proposed that they actually follow similar embryologic segments to hemangiomas (S1, S2, etc.). With either delineation, it seems that the involvement of the forehead and the more of the face involved (as long as the PWS includes some part of the V1/S1 distribution) is the highest risk. Therefore a PWS that covers the forehead, upper cheek, and lower cheek has a higher risk than a PWS that covers only the forehead. The leptomeningeal component of SWS is present in only 10% of patients with all or most of one V1 branch of the trigeminal nerve involved. If bilateral V1 or V1/V2/V3 are involved the rate of SWS is significantly higher. Leptomeningeal angiomatosis may clinically manifest as epilepsy, mental retardation, hemiplegia, hemisensory defects, and homonymous hemianopsia. Characteristic calcifications are present in the outer layers of the cerebral cortex; these consist of double-contoured “tram tracks” that follow the brain convolutions. The most important initial evaluation of an infant with a PWS that involves the eyelid is by an ophthalmologist to rule out congenital glaucoma because this can be vision threatening. Ocular abnormalities, such as glaucoma, buphthalmos (infantile glaucoma, related to abnormal development of angle formed by cornea and iris), retinal detachment, and blindness can occur in SWS. These may be present without leptomeningeal involvement.
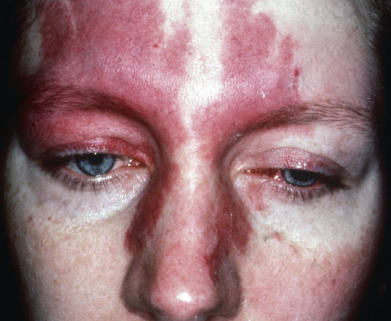
SWS results from the persistence of the primitive embryonal vascular plexus that develops during the sixth fetal week around the cephalic neural tube and in the region destined to become facial skin. Normally, the plexus regresses during the ninth week, but in SWS it persists. Mutations in GNAQ and GNA11 have been implicated, although it is unclear if the mutation alone is sufficient to cause SWS.
Because the risk of SWS is fairly low even with a PWS on the forehead, historically not all infants underwent imaging because the management did not necessarily change. It has now been demonstrated that early institution of aspirin may help prevent seizures, cognitive delays, and hemiparesis. Therefore early imaging to rule out SWS is warranted in patients who have a significant risk to be able to know whom to offer aspirin. Although calcifications are the most notable CNS finding and can be seen on computed tomography (CT) scan, they can develop later, so an MRI is the most sensitive test in infancy.
PiK3CA- Related Overgrowth Syndromes (PROS)
Mutations in PIK3CA cause multiple different syndromes with varied findings of CM with overgrowth. Depending on which mutation occurs and how much tissue is involved (due to mosaicism) patients can have various phenotypes: MCAP or CLOVES syndrome. Collectively, because there is some overlap, these (along with fibroadipose hyperplasia and hemimegalencephaly) are collectively known as PIK3CA -related overgrowth syndromes (PROS). Klippel-Trenaunay syndrome and KTS–Parkes-Weber syndrome are also caused by postzygotic mutations in PiK3CA. PIK3CA mutations have also recently been found in patients with CLAPO syndrome characterized by a CM of the lower lip and asymmetric overgrowth and head/neck lymphatic malformation.
Megalencephaly Capillary Malformation
MCAP was originally called M-CMTC and therefore CMTC was historically incorrectly associated with multiple congenital anomalies. The cutaneous lesions in MCAP are actually reticulated PWS (as described earlier) and not CMTC. Other features are an upper lip/nasal philtrum PWS, megalencephaly, a sandal toe deformity with 2-3 syndactyly of the toes, polymicrogyrisa, and skin elasticity. Patients should be followed for signs of hydrocephalus, and brain MRI is recommended. Renal ultrasound every 3 months until age 8 to rule out Wilms tumor has also been recommended.
CLOVES
The lipomatous overgrowth in CLOVES is typically very prominent at birth. Affected patients also often have large venous or venolymphatic malformations, as well as AVMs that can involved the skin but also importantly the CNS. Epidermal nevi can be localized or widespread. There is usually significant overgrowth of hands and/or feet. Patients can have spinal cord tethering in addition to renal agenesis. Patients require multidisciplinary care, including neurosurgery and orthopedic surgery and other specialties depending on their symptoms. Imaging to establish the presence of CNS vascular lesions or other anomalies helps guide therapy. Recently systemic rapamycin has been shown to be helpful in shrinking complicated vascular anomalies, including lymphatic malformations.
Klippel-Trenaunay Syndrome (KTS) and KTS–Parkes-Weber
Klippel-Trenaunay syndrome (KTS) is characterized as a triad of nevus flammeus, venous and lymphatic malformations, and soft tissue hypertrophy of the affected extremity ( Fig. 28.16 ). The lower limb is affected in approximately 95% of patients. When there is an associated arteriovenous (AV) fistula, Parkes-Weber is appended to the diagnosis.
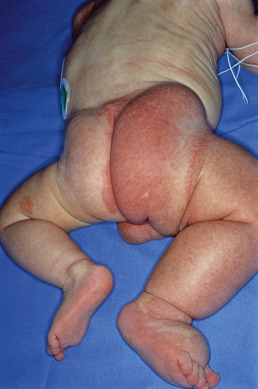
The earliest and most common presenting sign is a geometric CM. The deeper venous malformation (VM) in this sporadic syndrome may be confined to the skin, but it often extends to muscle and bone. The involved limb is usually larger and longer than normal. Venous thromboembolism has been reported, with an incidence as high as 22%. In other patients, the deep venous system is hypoplastic. Other, less frequent features include intermittent claudication, venous ulcers, increased skin temperature, diffuse hair loss, hypertrichosis, lymphedema, altered sweating, lacrimation, or salivation. Gait abnormalities are common. Intradural spinal cord AVMs, epidural hemangioma, and epidural angiomyolipoma have been reported to occur at the same segmental level as cutaneous lesions of KTS. Clinical evaluation consists of color duplex ultrasonography to evaluate the patency of the deep venous system, MRI for visualization of hypertrophic muscle and bone, arteriography when an AV fistula is suspected, and conventional radiography of both extremities. Early venography may be performed, if the deep venous system is not hypoplastic, to determine whether there are defects that might be amenable to surgical correction. When AV shunting is found the term KTS–Parkes-Weber is used.
Flashlamp-pumped pulsed dye laser treatments may be used for the nevus flammeus component. The varicosities and malformations may respond to microfoam sclerosis, endovenous thermal ablation, or surgical stripping. Edema is managed through elevation, graded compression pumps, fitted garments, and diuretics. Surgery may be performed to correct the inequality in limb length, to relieve deep venous obstruction, or to correct an associated AV fistula. Skin ulcers have responded to sunitinib. The Klippel-Trenaunay Support Group website can be found at www.k-t.org .
Diffuse Capillary Malformation with Overgrowth
Diffuse capillary malformation with overgrowth is a clinical phenotype proposed to describe patients with widespread PWS (often reticulated) with some usually nondebilitating proportionate overgrowth. Patients had normal neurologic function but require orthopedic follow-up for limb discrepancies.
Capillary Malformation–Arteriovenous Malformation
CM-AVM syndrome is an autosomal dominant disorder caused by heterozygous RASA1 mutations and resulting in multifocal CMs and high risk for fast-flow lesions. The CMs in CM-AVM may have arterial flow. Affected individuals have widespread “thumbprint” or café au lait–like oval CMs but can also have AVMs in the brain and spine. Therefore imaging of the brain and complete spine to rule out AVMs is recommended. Cobb syndrome (cutaneous meningospinal angiomatosis) is characterized by a port wine hemangioma or other vascular malformation in a dermatome supplied by a segment of the spinal cord containing a VM or AVM and may also be from RASA1 mutations.
Capillary Malformation–Arteriovenous Malformation 2
CM-AVM2 is a newly described similar phenotype caused by EPHRIN B4 mutation but an apparently lower risk of CNS AVMs.
Microcephaly Capillary Malformation Syndrome
Microcephaly capillary malformation syndrome is characterized by microcephaly with seizures and severe developmental delay, along with similar small discrete CMs as are seen in CM-AVM.
Proteus Syndrome
Proteus syndrome is characterized by vascular malformations that include nevus flammeus, hemihypertrophy, macrodactyly, verrucous epidermal nevus, soft tissue subcutaneous masses, and cerebriform overgrowth of the plantar surface. It is caused by an AKT-1 mutation and is reviewed in Chapter 27 .
Beckwith-Wiedemann Syndrome
Patients with Beckwith-Wiedemann syndrome have prominent nevus simplex especially on the glabella in addition to a protruding large tongue, posterior ear pits on the helix, omphalocele or other anterior abdominal wall defects, and hypoglycemia. Other rare syndromes associated with nevus simplex include Nova syndrome (patients have hydrocephalus), odontodysplasia (patients have defects in dentin and failed rupture of teeth), and Roberts-SC syndrome (symmetric limb defects and developmental delay).
TAR Syndrome
TAR syndrome is defined by congenital thrombocytopenia and bilateral absence or hypoplasia of the radius, and some patients have been reported to have port wine stain.
Adams DM, et al: Efficacy and safety of sirolimus in the treatment of complicated vascular anomalies. Pediatrics 2016; 137: e20153257.
Amyere M, et al: Germline loss-of-function mutations in EPHB4 cause a second form of capillary malformation-arteriovenous malformation (CM-AVM2) deregulating RAS-MAPK signaling. Circulation 2017; 136: 1037.
Cerrati EW, et al: Surgical treatment of head and neck port-wine stains by means of a staged zonal approach. Plast Reconstr Surg 2014; 134: 1003.
Griffin TD, et al: Port wine stain treated with a combination of pulsed dye laser and topical rapamycin ointment. Lasers Surg Med 2016; 48: 193.
Hackett CB, et al: Basal cell carcinoma of the ala nasi arising in a port wine stain treated using Mohs micrographic surgery and local flap reconstruction. Dermatolog Surg 2014; 40: 590.
Jagtap S, et al: Sturge-Weber syndrome. J Child Neurol 2013; 28: 725.
Kim C, et al: Histopathologic and ultrasound characteristics of cutaneous capillary malformations in a patient with capillary malformation–arteriovenous malformation syndrome. Pediatr Dermatol 2015; 32: 128.
Lacerda Lda S, et al: Differential diagnoses of overgrowth syndromes. Radiol Res Pract 2014; 2014: 947451.
Laquer VT, et al: Microarray analysis of port wine stains before and after pulsed dye laser treatment. Lasers Surg Med 2013; 45: 67.
Lee MS, et al: Diffuse capillary malformation with overgrowth. J Am Acad Dermatol 2013; 69: 589.
Lian CG, et al: Novel genetic mutations in a sporadic port-wine stain. JAMA Dermatol 2014; 150: 1336.
Martinez-Lopez A, et al: CLOVES syndrome. Clin Genet 2017; 91: 14.
McDonell LM, et al: Mutations in STAMBP, encoding a deubiquitinating enzyme, cause microcephaly–capillary malformation syndrome. Nat Genet 2013; 45: 556.
Memarzadeh A, et al: Limb length discrepancy in cutis marmorata telangiectatica congenita. Br J Dermatol 2014; 170: 681.
Mirzaa G, Conway R, Graham JM Jr, et al: PIK3CA-related segmental overgrowth. 2013 Aug 15. In: Adam MP, Ardinger HH, Pagon RA, et al. [Eds.] GeneReviews [Internet]. Seattle: University of Washington, Seattle.
Nguyen S, et al: Skin ulcers in Klippel-Trenaunay syndrome respond to sunitinib. Transl Res 2008; 151: 194.
Ortiz AE, et al: Port-wine stain laser treatments and novel approaches. Facial Plast Surg 2012; 28: 611.
Pleimes M, et al: Characteristic congenital reticular erythema. J Pediatr 2013; 163: 604.
Reddy KK, et al: Treatment of port-wine stains with a short pulse width 532-nm Nd:YAG laser. J Drugs Dermatol 2013; 12: 66.
Redondo P, et al: Microfoam treatment of Klippel-Trenaunay syndrome and vascular malformations. J Am Acad Dermatol 2008; 59: 355.
Rodriguez-Laguna L, et al: CLAPO syndrome. bioRxiv 2017; 154591.
Shirley MD, et al: Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med 2013; 368: 1971.
Swarr DT, et al: Expanding the differential diagnosis of fetal hydrops. Prenat Diagn 2013; 33: 1010.
Yiş U, et al: Capillary malformation–arteriovenous malformation syndrome with spinal involvement. Pediatr Dermatol 2014; 31: 744.
Venous Malformation
Venous malformations (VMs) present as rounded, blue or purple, spongy nodules. They often occur on the head and neck and may involve both the skin and the mucous membranes. There is usually a deep component with a connection to the venous circulation. Calcified phleboliths and localized hyperhidrosis may occasionally be present, and the lesions can sometimes be painful. In addition to typical VMs that are caused by a TEK mutation, there are other malformations categorized as VMs (glomuvenous malformations, cerebrocavernous malformations) Some lesions are amenable to sclerotherapy or surgical resection, but results are mixed. Compression may be helpful. Customized, snug-fitting garments are preferable to elastic bandages.
Several Syndromes Associated with Venous Malformations
Common and familial VMs in addition to blue rubber bleb nevus syndrome (BRBS) are caused by a TEK mutation ( TEK encodes the protein TIE-2). It is proposed that VMCM is due to a germline mutation and BRBS is caused by a somatic mutation (see following subsections). Patients with VMCM seem to have a germline mutation and then a second hit mutation in the TEK gene causes the VMs. In BRBS a progenitor cell may acquire a mutation in both TEK genes and then spread to other areas leading to multifocal lesions.
Multiple Cutaneous and Mucosal Venous Malformations (VMCM Syndrome)
VMCM patients have multiple small blue soft VMs. d -Dimer is sometimes elevated in VMCM but not in glomangiomas. The lesions of VMCM are small and unlike BRBS are not hyperkeratotic or firm.
Blue Rubber Bleb Nevus Syndrome
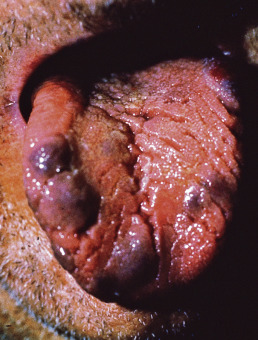
BRBS is characterized by cutaneous and GI venous malformations. There is often a dominant lesion that is much larger than the others. The skin lesions have a cyanotic, bluish appearance with a soft, elevated, nipple-like center, but deeper lesions may also occur. They can be emptied by firm pressure, leaving them flaccid. The lesions are located predominantly on the trunk and arms but also the palms and soles. Nocturnal pain may occur. Lesions can be found throughout the GI tract ( Fig. 28.17 ) but are numerous in the small intestine, and rupture of a lesion may produce melena. Occasionally, other organs may express VMs, and symptomatic CNS lesions have been described. Localized coagulopathy (DIC) can occur. Treatment of bleeding or painful lesions is destruction or excision. Minimally invasive surgical techniques are well suited to the treatment of numerous lesions. For patients who continue to have bleeding episodes that require blood transfusions, octreotide, a somatostatin analog known to decrease splanchnic blood flow, may be effective. ε-Aminocaproic acid has also been used.
Bannayan-Riley-Ruvalcaba Syndrome
Bannayan-Riley-Ruvalcaba syndrome is described later in this chapter.
Maffucci Syndrome
Maffucci syndrome is characterized by multiple vascular malformations with dyschondroplasia caused in most patients by mutations in IDH1 or IDH2. The dyschondroplasia is manifested by uneven bone growth as a result of the defects of ossification, with enchondromatosis that results in multiple and frequent fractures in the period of bone growth. During the prepubertal years, 1–2 cm nodules appear on the small bones of the hand or foot. Later, larger nodules, the enchondromas, appear on the long bones. Much later, similar lesions appear on the trunk. Sarcomatous degeneration occurs in 50% of patients. The distribution of the lesions is mostly unilateral. Multiple VMs of the skin and mucous membranes are present in this nonhereditary mesodermal dysplasia disorder. Lymphangiomas may also occur. Pigmentary changes, such as vitiligo and café au lait macules, have been noted. In Ollier disease, the enchondromatosis is present without the cutaneous abnormalities.
Cerebral Cavernous Malformations
Cerebral cavernomas are vascular malformations in the brain that may be inherited in an autosomal dominant manner. The causative mutations are in CCM1 (KRIT1), CCM2, or PDCD10. Cutaneous malformations are sometimes present, including hyperkeratotic cutaneous capillary VMs.
Glomuvenous Malformation
VM should be distinguished from glomuvenous malformation (GM, glomangioma). GM is made up of glomus cells and is caused by mutations in glomulin. GM can be pink at initial presentation but evolve to blue-black with a cobblestone appearance and minimal hyperkeratosis. Involvement of an extremity is typical, and the GMs are often painful if compressed because they are made of glomus cells. The lesion often shrinks with external pressure and is typically painful in the morning due to congestion. Increased pain may be noted at puberty, during menstruation, with pregnancy, or with oral contraceptives. Sclerotherapy is more effective in VM than in GM.
Glomus Tumor
The solitary glomus or neuromyoarterial tumor (also known as a solitary glomangioma) is most frequently a skin-colored or slightly dusky blue, firm nodule 1–20 mm in diameter. The characteristic location is subungual, but tumors may occur on the fingers and arms, or elsewhere. Subungual tumors show a bluish tinge through the translucent nail plate. The tumor is usually extremely tender, and paroxysmal pain occurs frequently. Sensitivity is likely to be present constantly, and when touched the tumor responds with severe radiating pain. However, nontender glomus tumors are encountered. Digital lesions are more common in women, and there is a male predominance of nondigital lesions. Hereditary multiple glomus tumors may represent an autosomal dominant mosaic trait and may be congenital. There appears to be an association between glomus tumor and neurofibromatosis. High-resolution MRI, high-resolution ultrasonography (5–9 MHz), and color duplex sonography may be used to define the limits of the tumor before surgery is undertaken. Progressive growth may lead to ulceration.
Histologically, glomus tumors contain numerous vascular lumina lined by a single layer of flattened endothelial cells. Peripheral to the endothelial cells are layers of glomus cells. Generally, these are round and arranged in distinct rows resembling strings of black pearls. Rarely, the cells have a somewhat spindled morphology. Multiple glomus tumors tend to have only one or two layers of glomus cells. Glomangiomyomas have a prominent muscularis media in addition to one or two layers of glomus cells. Both solitary and multiple glomus tumors are related to the arterial segment of the cutaneous glomus, the Sucquet-Hoyer canal. The glomus cells are modified vascular smooth muscle cells and stain with vimentin rather than desmin. Smooth muscle actin is often positive.
Treatment of solitary glomus tumors is best carried out by complete excision, which immediately produces relief from pain. The subungual tumors are most difficult to locate and eradicate because they are usually small, seldom more than a few millimeters in diameter.
Rare reports of glomangiosarcomas describe large, deeply located extremity lesions that consist of sarcomatous areas intermingled with areas of benign glomus tumor.
Amyere M, et al: Common somatic alterations identified in Maffucci syndrome by molecular karyotyping. Mol Syndromol 2014; 5: 259.
de Vos IJ, et al: Review of familial cerebral cavernous malformations and report of seven additional families. Am J Med Genet A 2017; 173: 338.
Fayad LM, et al: Venous malformations. Skeletal Radiol 2008; 37: 895.
Ham KW, et al: Glomus tumors. Arch Plast Surg 2013; 40: 392.
Harrison B, Sammer D: Glomus tumors and neurofibromatosis. Plast Reconstr Surg Glob Open 2014; 2: e214.
Soblet J, et al: Blue rubber bleb nevus (BRBN) syndrome is caused by somatic TEK (TIE2) mutations. J Investig Dermatol 2017; 137: 207.
Yanai T, et al: Immunohistochemical demonstration of cyclooxygenase-2 in glomus tumors. J Bone Joint Surg Am 2013; 95: 725.
Arteriovenous Fistulas
An arteriovenous fistula (AV) is a route from artery to vein, bypassing the capillary bed. AV fistulas may be congenital or acquired. Congenital AV fistulas occur mostly on the extremities and may be recognized, or at least suspected, in the presence of varicose veins, ulcerations, and CMs. They may occur internally as a component of Osler-Weber-Rendu disease (hereditary hemorrhagic telangiectasia). Acquired AV fistulas are usually the result of trauma ( Fig. 28.18 ) but may be created intentionally for hemodialysis access.
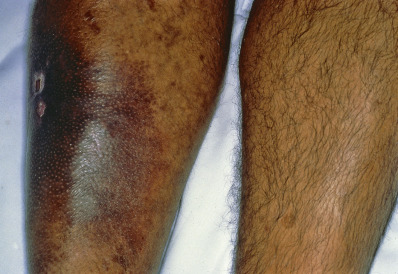
The skin over AV fistulas is warmer, hair may grow faster, and the affected limb may be larger than the other; thrills and bruits may be discerned in some cases. Changes may result from stasis, a vascular steal syndrome, edema, a vascular mass, increased sweating, or paresthesias. At times, reddish purple nodules or a plaque may be present with a clinical resemblance to Kaposi sarcoma; this has been called pseudo–Kaposi sarcoma (Stewart-Bluefarb syndrome). It may occur because of congenital malformations, in which case a unilateral purplish discoloration of the skin over or distal to the AV anomaly begins to appear in the second or third decade of life. This type accounts for 80% of cases; the remainder are secondary to fistulas caused by trauma. Iatrogenic AV fistulas, such as those produced to facilitate hemodialysis, may also bring about skin changes, including reactive angioendotheliomatosis. Histologically, there is an increase in thick-walled vessels lined by plump endothelial cells, extravasated erythrocytes, and deposits of hemosiderin. Proliferating endothelial cells may occlude the lumen.
Cirsoid aneurysms (angioma arteriale racemosum) are uncommon congenital AV fistulas of the scalp or face. They may appear on the skin as a pulsating mass that may extend over the neck and scalp and may penetrate into the cranium, or they may simply manifest as a solitary blue or red papule in the midadult period. Abdominal AV fistulas may be associated with lower extremity edema, cyanosis, pulsatile varicose veins, and scrotal edema.
Diagnosis of an AV fistula is established by plethysmography, thermography, determination of oxygen saturation of venous blood, or arteriography.
Treatment of traumatically induced AV fistulas by excision is curative. Because the congenital malformation variety consists of multiple small distal lesions, surgical intervention is not feasible in many patients. Color echo-Doppler ultrasonography–guided sclerotherapy with polidocanol microfoam has been used successfully in this setting. Sodium tetradecyl sulfate and ethanolamine oleate have both been used as sclerosants in various forms of AV malformation. Pressure and elevation as supportive measures may limit ulceration, infection, and other secondary complications.
Rosenberg TL, et al: Arteriovenous malformations of the head and neck. Otolaryngol Clin North Am 2018; 51: 185.
Rutherford RB: Noninvasive evaluation for congenital arteriovenous fistulas and malformations. Semin Vasc Surg 2012; 25: 49.
Scruggs J, et al: Cutaneous manifestations of abdominal arteriovenous fistulas. Cutis 2011; 87: 284.
Other Lesions
Nevus anemicus is a congenital disorder characterized by macules of varying size and shape that are paler than the surrounding skin ( Fig. 28.19 ) and cannot be made red by trauma, cold, or heat. The nevus resembles vitiligo, but there is a normal amount of melanin. Wood’s light does not accentuate it, and diascopy causes it to merge into the surrounding blanched skin. The patches are usually well defined with irregular edges. Nevus anemicus can be an important early marker of neurofibromatosis in a young child, before other stigmata have appeared. Nevus anemicus can also be found in tuberous sclerosis or as one component of phakomatosis pigmentovascularis. In nevus anemicus, a flare does not develop after rubbing the skin but does develop after rubbing the adjacent normal skin. The underlying defect is an increased sensitivity of the blood vessels to catecholamines. On biopsy and with confocal microscopy, lesional skin resembles normal skin.
Nevus oligemicus presents as a patch of livid skin that is cooler than the normal skin as a result of decreased blood flow. Vasoconstriction of deep vessels is thought to be the underlying defect.
Sinusoidal hemangioma is a vascular malformation that usually presents in adults as a bluish purple nodule, less than 4 cm in diameter, on the trunk or breasts. Multiple lesions may occur, and a facial location has also been reported. Histologically, it appears as a lobular, circumscribed mass with dilated, interconnected vascular channels filled with blood.
Ferrari F, et al: Juvenile xanthogranuloma and nevus anemicus in the diagnosis of neurofibromatosis type 1. JAMA Dermatol 2014; 150: 42.
Marque M, et al: Nevus anemicus in neurofibromatosis type 1. J Am Acad Dermatol 2013; 69: 768.
Prominent Inferior Labial Artery
The arteries supplying the lips are normally tortuous to accommodate the movements of the mouth. Howell and Freeman reported a potentially troublesome arterial anomaly of the lower lip characterized by the appearance of a pulsating papule in the lower vermilion, 1 or 2 cm from the oral commissure, formed by an especially tortuous segment of the inferior labial artery. A similar anomaly may involve the upper lip. Caliber-persistent labial artery may be misdiagnosed as squamous cell carcinoma, and the biopsy may produce significant bleeding. On the lip, it is best to “palpate for pulsation prior to puncture.”
Howell JB, Freeman RG: The potential peril from caliber-persistent arteries of the lips. J Am Acad Dermatol 2002; 46: 256.
Lymphatic Malformations
Microcystic Lymphatic Malformation (Lymphangioma Circumscriptum)
The old term for microcystic lymphatic malformation (MLM) was lymphangioma circumscriptum; however, this is not a tumor but rather a congenital malformation of the superficial lymphatics. An MLM presents as groups of deep-seated, vesicle-like papules ( Fig. 28.20 ), resembling frog spawn, at birth or shortly thereafter. The lesions are usually yellowish but may be pink, red, or dark when bled into. This creates the illusion that they are rapidly changing. When the papules are punctured, they exude clear, colorless lymph. The papules are arranged irregularly in groups that may be interconnected by sparsely scattered lymph cysts. The entire process, however, as a rule is localized to one region. The sites of predilection are the abdomen, axillae, genitalia, and mouth, particularly the tongue. The scrotum is subject to multifocal lymphatic malformations presenting as clear, thick-walled, vesicle lesions. At times, the surface is verrucous, in which case the color may be brownish, and the lesions may be mistaken for warts. Lesions resembling molluscum contagiosum have also been described.
Frequently, the lesions consist of a combination of blood and lymph elements, so that purple areas are sometimes seen scattered within the vesicle-like papules. The lesions are also frequently associated with a deep component that occupies the subcutaneous tissues and muscles. Over time, these lymphatic malformations show only slight changes.
As with angiokeratomas, lymphangiomas may be seen adjacent to café au lait macules. This may represent a twin spotting phenomenon. Acquired lesions occur in the setting of chronic lymphedema. Lesions occurring after radiation therapy overlap with atypical vascular lesion (AVL). A peculiar penicillamine-induced dermopathy may result from damage to the underlying supporting structures of the dermis and allow dilation of lymph vessels within areas of trauma, such as the dorsal hands and knees. Central facial involvement may be seen in variegate porphyria, and sites of chronic high-potency steroid application may develop lymphangiectasia.
Excision and grafting, fulguration, or coagulation is frequently unsatisfactory because of recurrences resulting from vascular connections between the surface lesions and deep-seated lymphatic cisterns. The deeper component should be evaluated by MRI or other suitable radiologic imaging to delineate the extent of deep involvement before planned procedures. Vaporization with the carbon dioxide (CO 2 ) laser may be successful if deeper components are not present. Pulsed dye laser, intense pulse light systems, sclerosants, and electrosurgical techniques have also been reported as effective. Keloid formation has been described after laser vaporization of genital lymphangiomas. Sclerotherapy has been reported as successful, and radiotherapy has been used successfully in select refractory cases.
Emer J, et al: A case of lymphangioma circumscriptum successfully treated with electrodessication following failure of pulsed dye laser. Dermatol Online J 2013; 19: 2.
Kupetsky EA, Pugliano-Mauro M: Lymphangioma circumscriptum. Dermatol Surg 2014; 40: 928.
Yang X, et al: Highly selective electrocoagulation therapy. Dermatol Surg 2014; 40: 899.
Macrocystic Lymphatic Malformation
Macrocystic lymphatic malformations are deep-seated, typically multilocular, poorly defined, soft tissue masses that are painless and covered by normal skin. They are most common in the oral cavity and on the extremities and have been described in Maffucci syndrome . Cystic hygromas are clinically better circumscribed, occurring usually in the neck ( Fig. 28.21 ), axilla, or groin. The posterior neck lesions may be associated with Turner syndrome, other chromosomal aneuploidy conditions, hydrops fetalis, or other congenital abnormalities. Cytogenic analysis of children born with cystic hygromas is indicated, because aneuploidy may recur in subsequent pregnancies. Transabdominal or transvaginal sonography can visualize these lesions in utero. Usually, the lesions will recur after surgical treatment because of their depth, but injection sclerotherapy with agents such as OK-432 (picibanil) may result in regression. Sildenafil has been reported as an effective nonsurgical treatment in the setting of pediatric orbital lymphangioma.
Gandhi NG, et al: Sildenafil for pediatric orbital lymphangioma. JAMA Ophthalmol 2013; 131: 1228.
Guruprasad Y, et al: Cervical cystic hygroma. J Maxillofac Oral Surg 2012; 11: 333.
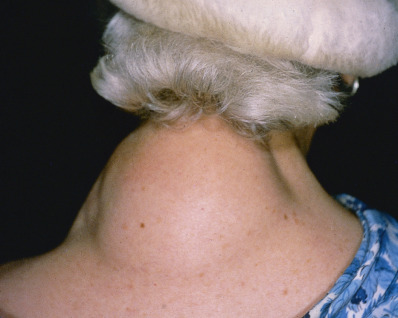
Lymphangiomatosis
Diffuse or multifocal dilated lymphatic channels involving the skin, soft tissues, bone, and parenchymal organs are a rare congenital condition. If an extremity is affected, the prognosis is good; however, when vital internal organs are involved, the prognosis is poor. Skin lesions are presenting signs in 7% of patients with thoracic lymphangiomatosis. These patients have a high incidence of complications, including chylothorax (49%), pulmonary infiltrates (45%), bone lesions (39%), splenic lesions (19%), cervical involvement (15%), and DIC (9%). Splenic lymphangiomatosis has been associated with Proteus syndrome. Diffuse pulmonary lymphangiomatosis has been successfully treated with bevacizumab.
Kaposiform Lymphangiomatosis
Kaposiform lymphangiomatosis is a newly described entity made of spindled lymphatic endothelial cells that grow progressively and is in between a tumor and a malformation. The intrathoracic disease is prominent and it can present in the skin with subcutaneous masses or ecchymosis. Histopathology shows spindled cells that are arranged in parallel, strands and sheets that anastomose, and is less well defined than KHE. Kasabach-Merritt–like coagulopathy has been reported. There is significant mortality but sirolimus therapy shows promise.
Gorham-Stout Syndrome
Gorham-Stout syndrome is characterized by lymphangiomatosis and chylous effusions, with osteolytic changes resulting in “vanishing bones.” The cutaneous lymphatic malformation may be the initial sign of the disease, which typically appears in young children, usually in areas adjacent to involved bones. If a patient has a lymphatic malformation that becomes painful, Gorham should be considered. Although multiple areas of the skeletal system may be involved, usually only a single bone is destroyed. The bone is completely or partially replaced with fibrous tissue. Sirolimus therapy has been helpful in some patients. Response to pegylated IFN alfa-2b was noted in a 9-year-old boy with systemic disease. Response to bisphosphonates has also been noted.
Multifocal Lymphangioendotheliomatosis
Patients with multifocal lymphangioendotheliomatosis present at birth with hundreds of red-brown plaques as large as several centimeters. Similar lesions occur in the GI tract and are associated with severe bleeding with a significant risk of mortality. Severe thrombocytopenia occurs in affected children with a coagulopathy (Kasabach-Merritt–like) that can be life threatening. There are reports of children with very few skin lesions and severe thrombocytopenia and reports of children with classic skin lesions but no thrombocytopenia. Treatment with corticosteroids and/or IFN alfa results in little to no improvement, but recently sirolimus has shown significant benefit. One patient was reported who did well with a liver transplant. The histology is distinctive, with delicate, thin-walled vessels lined by hobnailed endothelium with papillary tufting. The endothelial cells demonstrate a high proliferative fraction with Ki-67 staining and are reactive with LYVE-1, suggesting lymphatic differentiation.
Acquired Progressive Lymphangioma (Benign Lymphangioendothelioma)
This is a group of slow-growing lymphangiomas that present as bruiselike lesions or erythematous macules in children or middle-aged adults. Rarely, the lesion is yellow or alopecic. The histologic appearance is that of delicate, endothelium-lined spaces dissecting between collagen bundles. A similarity to the plaque stage of Kaposi sarcoma (KS) may be striking. Simple excision is curative. Topical and systemic steroids in addition to surgery have been effective.
Al-Jamali J, et al: Gorham-Stout syndrome of the facial bones. Oral Surg Oral Med Oral Pathol Oral Radiol 2012; 114: e23.
Aman J, et al: Successful treatment of diffuse pulmonary lymphangiomatosis with bevacizumab. Ann Intern Med 2012; 156: 839.
Croteau SE, et al: Kaposiform lymphangiomatosis. J Pediatr 2014; 164: 383.
Droitcourt C, et al: Multifocal lymphangioendotheliomatosis with thrombocytopenia. Pediatrics 2015; 136: e517.
Hagendoorn J, et al: Novel molecular pathways in Gorham disease. Pediatr Blood Cancer 2014; 61: 401.
Nikolaou VS, et al: Vanishing bone disease (Gorham-Stout syndrome). World J Orthop 2014; 5: 694.
Ricci KW et al: A phase 2 clinical trial assessing efficacy and safety of the mTOR inhibitor sirolimus in the treatment of generalized lymphatic anomaly, kaposiform lymphangiomatosis, and Gorham-Stout disease. Am J Respir Crit Care Med 2015; 191: A5454.
Salman A, et al: Acquired progressive lymphangioma. Pediatr Dermatol 2017; 34: e302.
Yang CH, et al: Orthotopic liver transplant for multifocal lymphangioendotheliomatosis with thrombocytopenia. Pediatr Transplant 2016; 20: 456.
Phakomatosis Pigmentovascularis
Patients with a combination of vascular malformations and melanocytic ( Fig. 28.22 ) or epidermal nevi are grouped into this disorder. The revised classification includes only four types: phakomatosis cesioflammea (blue nevus/dermal melanosis and nevus flammeus), phakomatosis spilorosa (nevus spilus and a pale-pink vascular spot), phakomatosis cesiomarmorata (blue spots and cutis marmorata telangiectatica congenita), and unclassifiable cases not corresponding to the previous patterns. These may all be phenotypes of a common disorder, and a recent report shows GNAQ or GNA11 mutations. Associated systemic findings may include intracranial and visceral vascular anomalies, ocular abnormalities, choroidal melanoma, and hemihypertrophy of the limbs. Phakomatosis cesioflammea is the most common type (85%), and half of patients with this type have serious manifestations. Bilateral deafness and malignant hypertension have also been described. Some authors have suggested that particularly extensive and aberrant mongolian spots may be a marker for more severe systemic involvement. Phakomatosis spilorosa has been associated with multiple granular cell tumors.
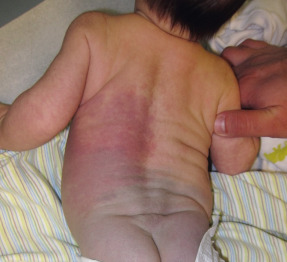
Phacomatosis pigmentokeratotica is now classified separately as a syndrome that includes speckled lentiginous nevi and nevus sebaceous and may be on the same spectrum as Schimmelpenning syndrome (nevus sebaceous syndrome). There can be associations of seizures, epibulbar dermoids, and potential malignancies. Phacomatosis pigmentokeratotica has been shown to be caused by an HRAS or KRAS mutation and can be considered a RASopathy (see Chapter 27 ).
Arnold AW, et al: Phacomatosis melanorosea without extracutaneous features. Eur J Dermatol 2012; 22: 473.
Groesser L, et al: Phacomatosis pigmentokeratotica is caused by a postzygotic HRAS mutation in a multipotent progenitor cell. J Investig Dermatol 2013; 133: 1998.
Om A, et al: Phacomatosis pigmentokeratotica. Pediatr Dermatol 2017; 34: 352.
Thomas AC, et al: Mosaic activating mutations in GNA11 and GNAQ are associated with phakomatosis pigmentovascularis and extensive dermal melanocytosis. J Investig Dermatol 2016; 136: 770.
Dilation of Preexisting Vessels
Spider Angioma (Vascular Spider, Spider Nevus, Nevus Araneus)
The lesion of spider angioma is suggestive of a red spider. The ascending central arteriole represents the “body” of the spider, and the radiating fine vessels suggest the multiple legs. These small telangiectases occur singly or severally, most frequently on the face, neck, and dorsal hands, indicating they may be induced by sun exposure.
Young children and pregnant women show these lesions most frequently. In pregnant women, palmar erythema is usually present with the vascular spiders. The presence of vascular spiders in otherwise healthy children is common.
Vascular spiders also occur in patients with cirrhosis, hepatitis C, malignant disease of the liver, and other hepatic dysfunctions. The common denominator has been shown to be an elevated blood estrogen level. Elevations in VEGF and basic fibroblastic growth factor are also significant predictors for spider angiomas in cirrhotic patients. When vascular spiders occur with palmar erythema and pallid nails with distal hyperemic bands, cirrhosis of the liver should be considered. AV hemangioma has also been reported to be associated with chronic liver disease.
The vascular spiders of childhood may involute without treatment, although several years may elapse before this occurs. In pregnant women, most lesions will involute soon after delivery. If active therapy will be performed pulse dye laser is effective without scarring, but electrodesiccation may produce good results in experienced hands.
Venous Lakes
Venous lakes (phlebectases) are small, dark-blue, slightly elevated blebs ( Fig. 28.23 ). They are easily compressed and are located on the face, ears, lips, neck, forearms, and backs of the hands. These manifestations of chronic sun damage are extremely dilated, blood-filled spaces lined with thin, elongated endothelial cells and usually surrounded by prominent solar elastosis. Venous lakes may be treated by light electrocautery, laser ablation, fulguration, infrared coagulation, intralesional injection of 1% polidocanol, and cryotherapy.
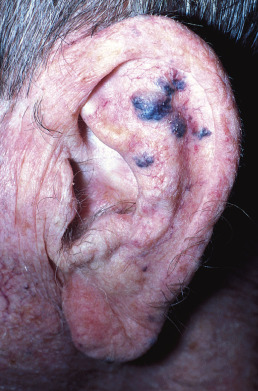
Capillary Aneurysms
These flesh-colored solitary lesions, resembling an intradermal nevus, may suddenly grow larger and darker and become blue-black or black as a result of thrombosis. Capillary aneurysms are surrounded by a zone of erythema. The lesions may be clinically indistinguishable from malignant melanoma. Histologically, these are thrombotic, dilated capillaries lying just below the epidermis. Shave excision in stages will expose the clot and eliminate the uncertainty.
Telangiectasia
Telangiectases are fine, linear vessels coursing on the surface of the skin; collectively, they are named telangiectasia. Telangiectasia may occur in normal skin at any age, in both genders, and can occur anywhere on the skin and mucous membranes but if abundant or with other symptoms, they can be important markers of systemic disease. Fine telangiectases may be seen on the alae nasi of most adults. They are prominent in areas of chronic actinic damage seen in fair-skinned persons. Persons long exposed to wind, cold, or heat are also subject to telangiectasia.
Telangiectases can be found in such conditions as radiodermatitis, xeroderma pigmentosum, lupus erythematosus (LE), dermatomyositis, scleroderma and the CREST syndrome, rosacea, cirrhosis of the liver, acquired immunodeficiency syndrome (AIDS), poikiloderma, basal cell carcinoma, necrobiosis lipoidica diabeticorum, sarcoid, lupus vulgaris, adenoma sebaceum, keloid, angioma serpiginosum, angiokeratoma corporis diffusum, ataxia-telangiectasia, pregnancy, and Bloom syndrome. These entities are discussed in other sections with the disease states in which they occur.
Altered capillary patterns on the fingernail folds (cuticular telangiectases) are indicative of collagen vascular disease, such as LE, scleroderma, or dermatomyositis. Tortuous glomeruloid loops are characteristic of LE, whereas dilated loops and avascular areas are typical of scleroderma and dermatomyositis. Reticular telangiectatic erythema may occur overlying implantable cardioverter-defibrillators.
Electrodessication and laser ablation can be effective. Pulsed dye laser and other vascular lasers, such as the 532-nm Nd:YAG laser, are usually well tolerated and associated with a low risk of scarring. Larger vessels require a longer pulse duration. Contact or cryospray cooling can reduce the incidence of complications. Pulse stacking (multiple pulses of low fluences) has been used to reduce the incidence of side effects, such as purpura, hyperpigmentation, hypopigmentation, and scar formation.
Hereditary Hemorrhagic Telangiectasia
Hereditary hemorrhagic telangiectasia (HHT, Osler-Weber-Rendu disease) is a genetic syndrome caused by mutations in either ENG, ACVRL1, GDF2, or SMAD4. The telangiectasias are widespread and usually involve the mucosa. They can be very tiny or much larger patches that can bleed easily. Patients often present with severe nose bleeds in childhood or GI bleeding less commonly. Patients with mutations in SMAD4 and have GI polyps. There are AVMs that can affect the skin, CNS, liver, GI tract, or lungs. Genetic testing can help prove the diagnosis and screening with pulmonary, CNS and liver imaging is recommended in affected patients to rule out AVMs in these locations. In addition to other causes of telangiectasias outlined earlier, CM-AVM and CM-AVM2 are in the differential.
Generalized Essential Telangiectasia
Generalized essential telangiectasia (GET) is characterized by the dilation of veins and capillaries over a large segment of the body without preceding or coexisting skin lesions. The telangiectases may be distributed over the entire body or localized to some large area, such as the legs, arms, and trunk. The lesions may be discrete or confluent. Distribution along the course of the cutaneous nerves may occur. This type of telangiectasia is rarely associated with systemic disease, although patients with a similar appearance may have autoimmune disease. One report documented GI bleeding from a “watermelon” stomach in a woman with GET.
Most frequently, GET develops in women in their forties and fifties. The initial onset is on the lower legs and then spreads to the upper legs, abdomen, and arms. The dilations persist indefinitely. Generally, this is a sporadic condition, although it has been described in families as an autosomal dominant trait, in which case it has been termed hereditary benign telangiectasia.
It has been reported that GET may be differentiated from telangiectasia associated with systemic disease by assessing alkaline phosphatase activity. Telangiectatic vessels in GET do not have alkaline phosphatase activity in the endothelium of the terminal arteriole and the arterial portion of the capillary loops.
Individual areas may be treated with laser ablation. High-energy, high-frequency, long-pulse Nd:YAG laser and the 585-nm flashlamp-pumped pulsed dye laser have been reported to produce good results. Tetracycline, ketoconazole, and treatment of a chronic sinus infection have led to involution in individual reports.
Unilateral Nevoid Telangiectasia
In unilateral nevoid telangiectasia, fine, threadlike telangiectases develop in a unilateral, sometimes dermatomal, distribution ( Fig. 28.24 ). The areas most often involved are the trigeminal and C3 and C4 or adjacent areas, with the right side involved slightly more often than the left. In some cases the condition is congenital, but more often it is acquired. Increased estrogen appears to play a role in the onset of acquired cases (e.g., pregnancy, puberty in women, adrenarche in men), and hepatitis/alcohol-related cases have been reported. Bilateral nevoid telangiectasia has been reported to be more common in older people, men and in the setting of some other illness such as liver disease or diabetes. Lesions have responded to pulsed dye laser treatment.