Raynaud Phenomenon and Raynaud Disease
Raynaud phenomenon is characterized by episodic, recurrent vasospasm of the fingers and toes resulting in white, blue, and red discoloration provoked by cold or stress. When it occurs in the presence of an associated disease, usually collagen vascular disease and often systemic sclerosis/scleroderma, it is called secondary Raynaud phenomenon. Raynaud disease (or primary Raynaud disease) occurs in the absence of associated illness. Although no significant structural changes occur in primary Raynaud disease, in secondary Raynaud phenomenon, especially when associated with connective tissue disease, sustained and recurrent vasospasm may lead to vessel wall damage.
In a series of 165 patients with Raynaud phenomenon, 51 had primary Raynaud disease. A defined connective tissue disease was present in about one third of the remaining patients, but 54 had undefined connective tissue disease (35 with positive antinuclear antibody [ANA] titer). In another study of 142 patients with idiopathic Raynaud phenomenon followed for more than 10 years, 14% progressed to a definite connective tissue disease. The initial presence of ANAs, thickening of fingers, older age at onset, and female gender were predictors of connective tissue disease. In a larger study of 3035 patients with primary Raynaud phenomenon, age of onset after 40 and progressively worsening Raynaud attacks were predictive of eventual diagnosis of a connective tissue disease (and reclassification as secondary Raynaud phenomenon), a development that occurred in 37.2% of patients after a mean of 4.8 years of follow-up. Sequential nailfold capillary microscopy and autoantibody determinations can predict development of systemic sclerosis in those with Raynaud phenomenon. The absence of nailfold capillaroscopic findings, conversely, predicts the presence of primary Raynaud disease (no associated systemic illness). Laser Doppler perfusion imaging or Doppler ultrasonography may enhance the evaluation of vascular damage from Raynaud disease. Technetium digital blood flow scintigraphy and skin temperature measurement of the fingers and toes by digital thermography may aid in the early diagnosis of Raynaud phenomenon of either the primary or secondary type.
Many of the studies on pathogenesis and therapy in Raynaud phenomenon are conducted on patients with systemic sclerosis/scleroderma, so it may not be possible to translate these findings to patients with primary Raynaud disease. However, cold exposure is a major trigger of vasospasm in all Raynaud patients. The exaggerated sympathetic response to cold may be caused by both excessive vasoconstrictor tone and a weak systemic vasodilation process, centrally mediated at least in part. The abnormal sympathetic response may also explain why some patients say that “stress” triggers Raynaud attacks. High homocysteine levels have been detected in patients with both primary and secondary Raynaud phenomenon. Patients with systemic sclerosis and Raynaud disease have elevated levels of endothelin 1 (ET-1), which correlates with both nailfold capillaroscopic findings and more advanced disease.
Secondary Raynaud Phenomenon
Raynaud phenomenon is produced by an intermittent constriction of the small digital arteries and arterioles. The digits have sequential pallor, cyanosis, and rubor. The involved parts are affected by ischemic paroxysms, which cause them to become pale, cold to the touch, and numb. The phenomenon is more frequently observed in cold weather. When exposed to cold, the digits become white (ischemic), then blue (cyanotic), and finally red (hyperemic). Over time, the parts may fail to regain their normal circulation between attacks and become persistently cyanotic and painful. If this phenomenon persists over a long period, punctate superficial necrosis of the fingertips develops; later, even gangrene may occur.
Secondary Raynaud phenomenon occurs most frequently in young to middle-aged women. It occurs with scleroderma, dermatomyositis, lupus erythematosus (LE, particularly those with anti-Sm and anti-RNP antibodies), mixed connective tissue disease, Sjögren syndrome, rheumatoid arthritis, and paroxysmal hemoglobinuria. Scleroderma was the underlying diagnosis in more than half of patients in one series. Occlusive arterial diseases, such as embolism, thromboangiitis obliterans, arteriosclerosis obliterans, and large-vessel vasculitis (Takayasu arteritis), may be present. In addition, various diseases of the nervous system, including cervical rib, scalenus anticus syndrome, and complex regional pain syndrome (reflex sympathetic dystrophy), may produce the disorder. Physical trauma, such as hand-transmitted vibration, as occurs with pneumatic hammer operation, can induce a syndrome identical to Raynaud and has been termed vibration white finger or hand-arm vibration syndrome. Pianists and typists may also develop this phenomenon. Raynaud phenomenon is a well-recognized complication after cold injury, especially frostbite. Pharmacologic agents, such as bleomycin, cisplatin-based chemotherapy, ergot, β-adrenergic blockers (including eye drops), cyclosporine, interferon (IFN)–α and IFN-β, vinyl polychloride exposure, and cocaine, may also be the cause. A French national pharmacovigilance database revealed 175 reports of Raynaud phenomenon among 307,128 adverse drug reports. Women were 61% of cases, and 8% of affected patients had a prior history of Raynaud. New agents reported included ribavirin, gemcitabine, hepatitis vaccine, isotretinoin, leflunomide, hydroxycarbamide, rofecoxib, telmisartan, and zolmitriptan. The clumping of red blood cells (RBCs) is believed to be responsible for the induction of Raynaud phenomenon, with high titers of circulating cold agglutinins. It may occur in cryoglobulinemia and polycythemia vera. Patients with cancer may develop Raynaud as a paraneoplastic phenomenon. Endocrine disorders, such as acromegaly, pheochromocytoma, carcinoid, and hypothyroidism, may present with or be associated with Raynaud phenomenon. Raynaud of the nipple is a variant of Raynaud disease that is difficult to diagnose. It presents with severe pain during lactation and must be distinguished from nipple candidiasis and eczema. Patients report the onset of symptoms during pregnancy and, when asked, will say that the symptoms are triggered by cold and accompanied by biphasic or triphasic color changes of the nipple. Nifedipine can be highly effective in this condition and is safe for use during lactation; minimal drug is found in the breast milk.
Simple tests and physical examination will generally distinguish Raynaud disease from secondary Raynaud phenomenon. Sclerodactyly, digital pitted scars, puffy fingers with telangiectasias, positive ANA, subcutaneous calcifications, basilar lung fibrosis, and changes on nailfold capillary microscopy (avascular “skip” areas with irregularly dilated capillary loops) are signs of connective tissue disease.
Raynaud Disease (Primary Raynaud Disease)
Raynaud disease is a primary disorder of cold sensitivity primarily seen in young women. The intermittent attacks of pallor, cyanosis, hyperemia, and numbness of the fingers are identical to those in secondary Raynaud phenomenon ( Fig. 35.1 ). The disease is usually bilateral, and gangrene occurs in less than 1% of cases.
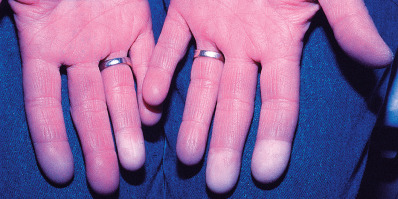
The diagnosis requires the absence of the diseases enumerated under secondary Raynaud phenomenon. An international panel proposed the following consensus criteria for differentiation of Raynaud disease from secondary Raynaud phenomenon: normal capillaroscopy; absence of physical findings suggestive of secondary causes, such as sclerodactyly, calcinosis, and ulcerations; no history of existing connective tissue disease; and negative or low-titer ANA (e.g., 1 : 40). Although some suggest that Raynaud disease should be present for 2 years before being classified as a primary process, it may take as long as 11 years for some systemic disorders to manifest. Overall, fewer than half of patients presenting with Raynaud symptoms will prove to have a connective tissue disease. The prognosis is good for patients with primary Raynaud disease.
Treatment
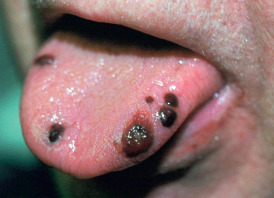
Treatments have often been studied but only in patients with secondary Raynaud phenomenon and digital ulceration associated with connective tissue disease, so not all treatments can be assumed to be effective in primary Raynaud disease or Raynaud secondary to other causes. If an underlying cause is found, treatment of the associated condition will often lead to improvement of Raynaud phenomenon. In both primary and secondary Raynaud, exposure to cold should be avoided. This includes avoidance of exposure to cold not only of the extremities but also of other parts of the body, because vasospasm may be induced by reduction of core body temperature, and Raynaud attack of atypical sites, such as the tongue, may occur. Warm gloves should be worn whenever possible. Trauma to the fingertips should be avoided. Smoking is absolutely contraindicated. A Raynaud attack may be broken at times by swinging the affected arm in a wide circle from the shoulder—the “windmill” maneuver. The use of standard nitroglycerin paste has had minimal efficacy and can produce systemic side effects. A new form of topical nitroglycerin, MQX-503, significantly improved skin blood flow without serious adverse events in a recent randomized controlled trial (RCT). Alternative treatments, including ginkgo and other herbal medications, have limited efficacy compared with the standard treatments and cannot be recommended for patients with significantly symptomatic disease.
Calcium channel blockers are the first-line therapy used in Raynaud disease because of their efficacy and low side-effect profile. Prolonged-release amlodipine or nifedipine is usually recommended. Some studies indicate that up to two thirds of treated patients will respond favorably. However, a Cochrane review of RCTs of calcium channel blockers for primary Raynaud disease concluded that their benefit was minimal, translating to 1.72 fewer attacks per week compared with placebo. Sildenafil and other phosphodiesterase-5 inhibitors are moderately effective in reducing Raynaud severity score, as well as the frequency and duration of Raynaud episodes, and may improve digital ulcer healing. These have become the second-line agents of choice. The angiotensin receptor antagonist losartan reduced the frequency and severity of attacks to a greater extent than nifedipine in an RCT. Conversely, angiotensin-converting enzyme (ACE) inhibitors failed to show benefit in an RCT and are therefore not recommended. Data on selective serotonin reuptake inhibitors (SSRIs; fluoxetine or ketanserin) are mixed, but SSRIs may be useful in refractory cases or when other agents are not tolerated. Intravenous (IV) biweekly N -acetylcysteine was effective in reducing the number of attacks in an observational study, relatively free of side effects. The use of statins, specifically atorvastatin, in patients with Raynaud caused by systemic sclerosis/scleroderma was associated with a reduction in Raynaud-associated symptoms, possibly through the vasoprotective actions of statins. Statin administration was associated with reduced circulating markers of vascular injury, which are usually elevated in scleroderma patients. Bosentan, an endothelin receptor (ETA and ETB) antagonist, significantly reduces the frequency of Raynaud attacks and reduces new digital ulcers. Iloprost, a prostaglandin analog, has substantial efficacy in scleroderma-associated Raynaud disease and digital ulceration, but it is only slightly better than nifedipine and significantly more expensive. Oral prostaglandins appear to lack similar efficacy, except perhaps at high doses.
In cases refractory to these medical treatments, surgical modalities can be considered. Botulinum toxin injections in the palm around each involved neurovascular bundle may lead to dramatic and at times immediate pain reduction. Ulcerations of the affected digits heal after the injections. The duration of response is often months to years, and injections can be repeated with similar efficacy. A review of 10 papers reporting 128 patients in the literature revealed 75%–100% improved, with ulcer healing in 75%–100% of reported patients, with 14.1% experiencing temporary hand weakness. Fat grafting or fat transfer to the hand has also been reported effective in a pilot study. Local digital sympathectomy can be effective and avoids amputation of chronically ulcerated digits. Cervical sympathectomy and endoscopic thoracic sympathectomy may give initial relief, but Raynaud symptoms often recur after 12–18 months. However, despite the return of symptoms, digital ulceration is greatly reduced. Compensatory hyperhidrosis is a common complication of thoracic sympathectomy.
Bank J, et al: Fat grafting to the hand in patients with Raynaud phenomenon. Plast Reconstr Surg 2014; 133: 1109.
Barrett ME, et al: Raynaud phenomenon of the nipple in breastfeeding mothers. JAMA Dermatol 2013; 149: 300.
Blagojevic J, Matucci-Cerinic M: Are statins useful for treating vascular involvement in systemic sclerosis? Nat Clin Pract Rheumatol 2009; 5: 70.
Boulon C, et al: Letter by Boulon and Constans regarding article “Relation of nailfold capillaries and autoantibodies to mortality in patients with Raynaud phenomenon.” Circulation 2016; 133: e668.
Bouquet E, et al: Unexpected drug-induced Raynaud phenomenon. Therapie 2017; 72: 547.
Bovenzi M: A longitudinal study of vibration white finger, cold response of digital arteries, and measure of daily vibration exposure. Int Arch Occup Environ Health 2009; 83: 259.
Caglayan E, et al: Vardenafil for the treatment of Raynaud phenomenon. Arch Intern Med 2012; 172: 1182.
Cappelli L, et al: Management of Raynaud phenomenon and digital ulcers in scleroderma. Rheum Dis Clin North Am 2015; 41: 419.
Cohen JC, et al: Raynaud’s phenomenon of the tongue. J Rheumatol 2013; 40: 336.
Coveliers HM, et al: Thoracic sympathectomy for digital ischemia. J Vasc Surg 2011; 54: 273.
Cutolo M, et al: Long-term treatment with endothelin receptor antagonist bosentan and iloprost improves fingertip blood perfusion in systemic sclerosis. J Rheumatol 2014; 41: 881.
De Angelis R, et al: Raynaud’s phenomenon. Clin Rheumatol 2003; 22: 279.
Ennis H, et al: Calcium channel blockers for primary Raynaud’s phenomenon. Cochrane Database Syst Rev 2014; 1: CD002069.
Funauchi M, et al: Effects of bosentan on the skin lesions. Rheumatol Int 2009; 29: 769.
Gargh K, et al: A retrospective clinical analysis of pharmacological modalities used for symptomatic relief of Raynaud’s phenomenon in children treated in a UK paediatric rheumatology centre. Rheumatology (Oxford) 2010; 49: 193.
Gliddon AE, et al: Prevention of vascular damage in scleroderma and autoimmune Raynaud’s phenomenon. Arthritis Rheum 2007; 56: 3837.
Goundry B, et al: Diagnosis and management of Raynaud’s phenomenon. BMJ 2012; 344: e289.
Herrick AL: Modified-release sildenafil reduces Raynaud’s phenomenon attack frequency in limited cutaneous systemic sclerosis. Arthritis Rheum 2011; 63: 775.
Hummers LK, et al: A multi-centre, blinded, randomised, placebo-controlled, laboratory-based study of MQX-503, a novel topical gel formulation of nitroglycerine, in patients with Raynaud phenomenon. Ann Rheum Dis 2013; 72: 1962.
Jenkins SN, et al: A pilot study evaluating the efficacy of botulinum toxin A in the treatment of Raynaud phenomenon. J Am Acad Dermatol 2013; 69: 834.
Koenig M, et al: Autoantibodies and microvascular damage are independent predictive factors for the progression of Raynaud’s phenomenon to systemic sclerosis. Arthritis Rheum 2008; 58: 3902.
Kowal-Bielecka O, et al: EULAR recommendations for the treatment of systemic sclerosis. Ann Rheum Dis 2009; 68: 620.
Lambova SN, Muller-Ladner U: The role of capillaroscopy in differentiation of primary and secondary Raynaud’s phenomenon in rheumatic diseases. Rheumatol Int 2009; 29: 1263.
Lim MJ, et al: Digital thermography of the fingers and toes in Raynaud’s phenomenon. J Korean Med Sci 2014; 29: 502.
Maverakis E, et al: International consensus criteria for the diagnosis of Raynaud’s phenomenon. J Autoimmun 2014; 48-49: 60.
Merritt WH: Role and rationale for extended periarterial sympathectomy in the management of severe Raynaud syndrome. Hand Clin 2015; 31: 101.
Mohokum M, et al: The association of Raynaud’s syndrome with cisplatin-based chemotherapy. Eur J Intern Med 2012; 23: 594.
Mondelli M, et al: Sympathetic skin response in primary Raynaud’s phenomenon. Clin Auton Res 2009; 19: 355.
Mueller M, et al: Relation of nailfold capillaries and autoantibodies to mortality in patients with Raynaud phenomenon. Circulation 2016; 133: 509.
Nagy Z, et al: Nailfold digital capillaroscopy in 447 patients with connective tissue disease and Raynaud’s disease. J Eur Acad Dermatol Venereol 2004; 18: 62.
Neumeister MW, et al: Botox therapy for ischemic digits. Plast Reconstr Surg 2009; 124: 191.
Pavlidis L, et al: Fat grafting to the hand in patients with Raynaud phenomenon. Plast Reconstr Surg 2015; 135: 229e.
Pavlov-Dolijanovic S, et al: Late appearance and exacerbation of primary Raynaud’s phenomenon attacks can predict future development of connective tissue disease. Rheumatol Int 2013; 33: 921.
Pope J, et al: Iloprost and cisaprost for Raynaud’s phenomenon in progressive systemic sclerosis. Cochrane Database Syst Rev 2000; 2: CD000953.
Prete M, et al: Raynaud’s phenomenon. Autoimmun Rev 2014; 13: 655.
Rosato E, et al: Laser Doppler perfusion imaging is useful in the study of Raynaud’s phenomenon and improves the capillaroscopic diagnosis. J Rheumatol 2009; 36: 2257.
Schiopu E, et al: Randomized placebo-controlled crossover trial of tadalafil in Raynaud’s phenomenon secondary to systemic sclerosis. J Rheumatol 2009; 36: 2264.
Segreto F, et al: The role of botulinum toxin A in the treatment of Raynaud phenomenon. Ann Plast Surg 2016; 77: 318.
Stewart M, Morling JR: Oral vasodilators for primary Raynaud’s phenomenon. Cochrane Database Syst Rev 2012; 7: CD006687.
Sunderkotter C, et al: Comparison of patients with and without digital ulcers in systemic sclerosis. Br J Dermatol 2009; 160: 835.
Erythromelalgia
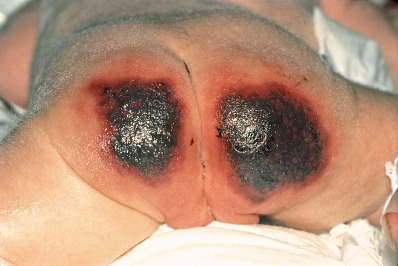
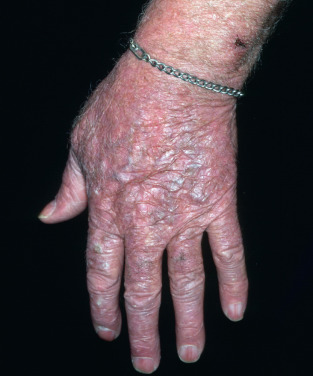
Also called erythermalgia and acromelalgia, erythromelalgia has a population-based incidence of 1.3 per 100,000 per year with a female predominance: 2.0 per 100,000 in women and 0.6 per 100,000 in men. Erythromelalgia is characterized by paroxysmal vasodilation of the feet, with burning, localized pain, redness, and high skin temperature. Infrequently, the hands ( Fig. 35.2 ), face, and ears may be involved. The burning paroxysms may last from a few minutes to several days and are usually triggered by an increase in environmental temperature or by exercise. The average patient has 1–2 attacks per week, but in some patients the attacks are much more frequent. Cooling and limb elevation can reduce the symptoms, but often relief can only be obtained by immersing the burning feet in ice water. More than 20% of patients will have evidence of cold injury, and more than 1% will have gangrene or undergo amputation. Quality of life is severely affected by the condition.
Erythromelalgia can be considered primary, secondary, or familial. For treatment purposes, secondary cases of erythromelalgia should be carefully divided into those associated with myeloproliferative diseases, often with elevated platelet counts, and other disorders. Myeloproliferative diseases associated with erythromelalgia include polycythemia vera, thrombotic thrombocytopenic purpura (TTP), and various forms of thrombocythemia. Administration of romiplostim, a thrombopoiesis-stimulating protein, has resulted in erythromelalgia. Low-dose aspirin is effective therapy for erythromelalgia associated with platelet abnormalities. If this fails, other methods to reduce the platelet count, such as administration of hydroxyurea, should be considered.
Acquired erythromelalgia has been reported secondary to topical exposure to isopropyl alcohol and after mushroom poisoning with Clitocybe acromelalga and Clitocybe amoenolens. Medications that have induced erythromelalgia include calcium channel blockers (both nifedipine and verapamil), ergot derivatives such as bromocriptine and pergolide, and cyclosporine. There may be a long period of treatment (years) with these agents before the appearance of the erythromelalgia. Chronic vibration and tobacco use have also been suggested as possible risk factors. Stopping the medication usually leads to improvement of symptoms within weeks.
In the vast majority of cases seen by dermatologists, erythromelalgia is probably a neurologic disorder. It can be seen in various neurologic conditions or diseases associated with neurologic sequelae, such as peripheral neuropathy, myelitis, multiple sclerosis, autoimmune small-fiber axonopathy, or diabetes mellitus. Erythromelalgia is sometimes associated with Raynaud phenomenon; both are disorders of abnormal neurovascular function. One series of 46 patients described concurrent Raynaud in 80% of patients. In many patients, no associated neurologic disease may be detected by routine neurologic examination, but careful neurologic testing will reveal evidence of a small-fiber neuropathy in the majority of such cases.
Inherited, familial, or hereditary erythromelalgia usually has its onset in childhood or adolescence (early or late onset). Familial cases have an autosomal dominant inheritance pattern. Familial erythromelalgia is now known to be an “inherited neuronal ion channelopathy.” The mutation is in the gene SCN9A, which encodes a peripheral sodium channel Na V 1.7. This is a mainly peripheral sodium channel with robust expression in dorsal root ganglion neurons and sympathetic ganglion neurons, especially those with nociceptive function. This sodium channel acts as a “threshold” channel and sets the gain in nociceptors. Many mutations in the affected gene have been mapped. Gene mutations causing erythromelalgia occur in areas that affect the structure of the actual channel by substituting amino acids in this critical location. The mutations causing erythromelalgia are gain-of-function mutations that lead to a hyperpolarizing shift of activation, allowing Na V 1.7 to open at lower potentials, enhancing excitability. Furthermore, high temperatures have been shown in vitro to cause a significant depolarizing shift in the mutant channels. Mutational analysis of one patient with careful nerve signaling evaluation demonstrated increased activity-dependent slowing of C-fibers. Mutational-guided therapy was used in two other patients with gain-of-function mutations in Na V 1.7, with carbamazepine attenuating the pain in both cases. Novel drugs such as PF-05089771 and TV-45070 have Na V 1.7 selectivity and may be promising future agents for patients with primary erythromelalgia.
The amount of gain of function correlates with age of onset of the disease; more significant mutations have earlier onset. The nature of the mutation also affects the binding of medications to the channel, so various mutations may have different responses to the same medication, depending on whether that mutation allows the drug to bind to the channel. Other gain-of-function mutations in SCN9A cause “paroxysmal extreme pain disorder” (formerly called familial rectal pain syndrome). This disorder has prominent autonomic manifestations that include skin flushing, sometimes with only half the face turning red (harlequin color change), syncope with bradycardia, and severe burning pain, most often rectal, ocular, or mandibular. One mutation in Na V 1.7 produced a clinical syndrome with features of both erythromelalgia and paroxysmal extreme pain disorder. Autosomal recessive nonsense mutations that cause loss of function of the Na V 1.7 channel result in the inability to sense pain. These patients are otherwise neurologically normal.
Interestingly, the association of secondary erythromelalgia with autoimmune conditions has led to the supposition that autoantibodies to the Na V 1.7 sodium channel may be present. Immunomodulatory therapy such as intravenous immune globulin (IVIG) has been used successfully in some patients with autoimmune disease and erythromelalgia. When severe, erythromelalgia is a life-altering disease, and aggressive management is warranted. Patients may benefit from referral to special clinics for pain management or pain rehabilitation. At times, simple measures such as immersion in cool water may stop pain crises. Biofeedback can be of benefit. In general, no more than 50% of patients with erythromelalgia of the neuropathic type will respond to any one medication, so the treatment must be tailored to each patient, and often combinations of agents are used. Topical amitriptyline 1% and ketamine 0.5% in a gel are safe topical options and are especially reasonable for affected thin-skinned areas, such as the face or ears, where penetration would be optimal. Topical midodrine 0.2% has been used successfully. Oral amitriptyline, sertraline, nortriptyline, pregabalin, and venlafaxine have shown benefit in some patients. Mexiletine, which has a normalizing effect on pathologic gating properties of the Na V 1.7 sodium channel mutation, has been reported effective in some patients, as have carbamazepine and the combination of carbamazepine and gabapentin. Corticosteroids have been used in select patients with erythromelalgia, and in one series appeared to be beneficial if used early, following a clear trigger (infectious, traumatic, or surgical) at the onset of erythromelalgia. Neurosurgical intervention has been used in the most severely affected, carefully selected patients who have failed medical management.
Red Ear Syndrome
Red ear syndrome describes a rarely reported disorder characterized by relapsing attacks of redness and burning affecting both ears, usually only one ear at a time. The attacks are more common in the winter and are precipitated by touching, movements, and exposure to warmth. Associated conditions include neural disorders of the trigeminal and glossopharyngeal nerves, migraines, and LE. It is unclear if red ear syndrome is a disease sui generis or is actually erythromelalgia of the ears. Treatment with oral and topical tricyclic antidepressants has been beneficial. Red ear syndrome must be distinguished from the springtime variant of polymorphous light eruption seen in young males with cold exposure, relapsing polychondritis (the lobe is also involved in red ear syndrome), cellulitis, and borrelial lymphocytoma.
Alhadad A, et al: Erythromelalgia. Vasa 2012; 41: 43.
Catterall WA, et al: Inherited neuronal ion channelopathies. J Neurosci 2008; 28: 11768.
Cerci FB, et al: Intractable erythromelalgia of the lower extremities successfully treated with lumbar sympathetic block. J Am Acad Dermatol 2013; 69: e270.
Chen MC, et al: Erythema associated with pain and warmth on face and ears. J Headache Pain 2014; 15: 18.
Cheng X, et al: Mutation l136V alters electrophysiological properties of the Na(v)1.7 channel in a family with onset of erythromelalgia in the second decade. Mol Pain 2008; 4: 1.
Cook-Norris RH, et al: Pediatric erythromelalgia. J Am Acad Dermatol 2012; 66: 416.
Cregg R, et al: Mexiletine as a treatment for primary erythromelalgia. Br J Pharmacol 2014; 171: 4455.
Crunkhorn S: Pain: blocking pain in inherited erythromelalgia. Nat Rev Drug Discov 2016; 15: 384.
David MD, et al: Topically applied midodrine 0.2%, an α1-agonist, for the treatment of erythromelalgia. JAMA Dermatol 2015; 151: 1025.
Drenth JP, et al: Primary erythermalgia as a sodium channelopathy. Arch Dermatol 2008; 144: 320.
Eberhardt M, et al: Inherited pain: sodium channel Na V 1.7 A1632T mutation causes erythromelalgia due to a shift of fast inactivation. J Biol Chem 2014; 289: 1971.
Eismann R, et al: Red ear syndrome. Dermatology 2011; 223: 196.
Estacion M, et al: Na V 1.7 gain-of-function mutations as a continuum. J Neurosci 2008; 28: 11079.
Fisher TZ, et al: A novel Na V 1.7 mutation producing carbamazepine-responsive erythromelalgia. Ann Neurol 2009; 65: 733.
Geha P, et al: Pharmacotherapy for pain in a family with inherited erythromelalgia guided by genomic analysis and functional profiling. JAMA Neurol 2016; 73: 659.
Genebriera J, et al: Results of computer-assisted sensory evaluation in 41 patients with erythromelalgia. Clin Exp Dermatol 2012; 37: 350.
Han JH, et al: Paraneoplastic erythromelalgia associated with breast carcinoma. Int J Dermatol 2012; 51: 878.
Iqbal J, et al: Experience with oral mexiletine in primary erythromelalgia in children. Ann Saudi Med 2009; 29: 316.
Kalava K, et al: Response of primary erythromelalgia to pregabalin therapy. J Clin Rheumatol 2013; 19: 284.
Kalgaard OM, et al: Prostacyclin reduces symptoms and sympathetic dysfunction in erythromelalgia in a double-blind randomized pilot study. Acta Derm Venereol 2003; 83: 442.
Kist AM, et al: SCN10A mutation in a patient with erythromelalgia enhances C-fiber activity dependent slowing. PLoS One 2016; 11: e0161789.
Kluger N, et al: Romiplostim-induced erythromelalgia in a patient with idiopathic thrombocytopenic purpura. Br J Dermatol 2009; 161: 482.
Lambru G, et al: The red ear syndrome. J Headache Pain 2013; 14: 83.
Lin KH, et al: Effectiveness of botulinum toxin A in treatment of refractory erythromelalgia. J Chin Med Assoc 2013; 76: 296.
Moody S, et al: Secondary erythromelalgia successfully treated with intravenous immunoglobulin. J Child Neurol 2012; 27: 922.
Nanayakkara PWB, et al: Verapamil-induced erythermalgia. Neth J Med 2007; 65: 349.
Natkunarajah J, et al: Treatment with carbamazepine and gabapentin of a patient with primary erythermalgia (erythromelalgia) identified to have a mutation in the SCN9A gene, encoding a voltage-gated sodium channel. Clin Exp Dermatol 2009; 34: e640.
Pagani-Estevez GL, et al: Erythromelalgia. J Am Acad Dermatol 2017; 76: 506.
Parker LK, et al: Clinical features and management of erythromelalgia. Clin Exp Rheumatol 2017; 35: 80.
Patel M, et al: Facial erythromelalgia. J Am Acad Dermatol 2014; 71: e250.
Pipili C, Cholongitas E: Erythromelalgia in a diabetic patient managed with gabapentin. Diabetes Res Clin Pract 2008; 79: e15.
Poterucha TJ, et al: Topical amitriptyline combined with ketamine for the treatment of erythromelalgia. J Drugs Dermatol 2013; 12: 308.
Raieli V, et al: Prevalence of red ear syndrome in juvenile primary headaches. Cephalalgia 2011; 31: 597.
Reed KB, Davis MDP: Incidence of erythromelalgia. J Eur Acad Dermatol Venereol 2009; 23: 13.
Ryan S, et al: Red ear syndrome. Cephalalgia 2013; 33: 190.
Tang Z, et al: Primary erythromelalgia. Orphanet J Rare Dis 2015; 10: 127.
Young FB: When adaptive processes go awry: gain-of-function in SCN9A. Clin Genet 2008; 73: 34.
Livedo Reticularis, Livedo Racemosa
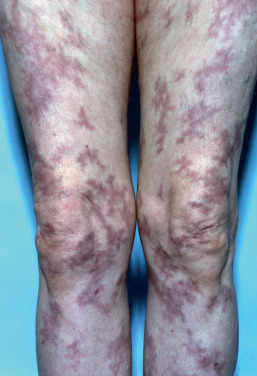
Livedo reticularis is the term used to describe a netlike, mottled or reticulated, pink or reddish blue discoloration of the skin, mostly on the extremities, especially the legs. It is more prominent with exposure to cold and may vanish with warming. It is usually seen on the lower extremities in young children and women. The pathogenic basis is reduced blood flow and lowered oxygen tension in the venous plexus of the skin. Cutis marmorata is another name for livedoid physiologic mottling of skin exposed to cold. For clinical purposes, it is best to separate livedo reticularis (a benign condition in most cases) from fixed livedo reticularis, better known as livedo racemosa. Livedo racemosa forms irregular networks and broken circular segments that are fixed and do not vary appreciably with temperature changes ( Fig. 35.3 ). The lesions are usually asymptomatic. If necrosis or purpura occurs over the livedoid areas, the terms necrotizing livedo and retiform purpura, respectively, may be used. Livedo racemosa and livedo with purpura or necrosis are almost always associated with significant systemic disease that requires treatment. Unfortunately, the literature does not always accurately separate these entities, and patients may present with variable livedo (resembling livedo reticularis) and later develop more fixed lesions. In addition, some patients who have more variable livedo may have serious underlying disease that may require evaluation and treatment. These patients may not be easily identifiable initially on physical examination features alone. In this section, the term livedo will be used to describe this cutaneous finding and its association with other conditions. When livedo reticularis is seen, the clinician should consider the following categories of diseases as possibly causal: physiologic, hypercoagulable states (including myelodysplasias, cancer, and antiphospholipid, cryoglobulinemia, and Sneddon syndromes), vasculitis (especially medium and large vessel), emboli, medications, and neurologic disorders.
Drugs may cause livedo. Amantadine (Symmetrel) can cause livedo reticularis. Quinidine and quinine may be associated with a photosensitivity that is livedoid in appearance, but on biopsy an interface dermatitis will be present. Minocycline can cause livedo, and this is a marker for the development of an antineutrophil cytoplasmic antibody (ANCA)–positive vasculitis in these patients. The medication must be stopped immediately. Other medications associated with livedo include gemcitabine, heparin (perhaps associated with heparin-induced antiplatelet antibodies), IFN-β, and bismuth. Cholesterol emboli after intravascular procedures or surgery may present with livedo, often in the setting of renal injury and eosinophilia (see later). The sheaths used for intravascular procedures may be coated with a hydrophilic polymer that may embolize and cause livedo (histopathology can reveal the polymer in the vessels).
Neurologic disorders can create livedo reticularis by altering innervation and, consequently, blood flow in the skin. Brain injury, multiple sclerosis, diabetes mellitus, poliomyelitis, and Parkinson disease are some examples.
Many of the syndromes with fixed livedo (livedo racemosa) have important systemic implications. These conditions can be either primary thrombotic processes or vascular inflammatory processes. It is important to consider in each case that if the vessels of the skin are affected, the vessels in other organs, specifically the central nervous system (CNS) and kidneys, may also be affected. Sneddon syndrome is a rare condition that occurs in young to middle-aged women. These patients present with livedo, report a history of migraines, and then develop cerebrovascular infarcts. The prognosis is poor. Frequently, patients have antiphospholipid antibodies (up to 85%) and may have enough features to be diagnosed with systemic lupus erythematosus (SLE). They would be accurately diagnosed as having antiphospholipid antibody syndrome. Other connective tissue diseases, such as dermatomyositis, rheumatoid arthritis, and systemic sclerosis, may have antiphospholipid antibodies and thus feature livedo. For this reason, patients with SLE and livedo are likely to have more severe disease manifestations, such as renal disease, vasculitis, and antiphospholipid antibodies, even in the absence of full-blown Sneddon syndrome. Headache may be the presenting symptom in these patients, and the misdiagnosis of migraine may initially be considered. Not all patients with Sneddon syndrome can be diagnosed as having antiphospholipid antibody syndrome, however, and their optimal evaluation and management is unclear. Other significant disorders with livedo as a skin manifestation include thrombotic processes (hypercoagulable states, type I cryoglobulinemia), microangiopathic hemolytic anemias (TTP, hemolytic uremic syndrome, disseminated intravascular coagulation), medium- and large-vessel vasculitides, and septicemia. Moyamoya disease is a rare, chronic cerebrovascular occlusive condition characterized by progressive stenosis of the arteries in the circle of Willis. Patients present with ischemic strokes or cerebral hemorrhages. Both idiopathic moyamoya disease and disease connected with factor V Leiden mutation have been associated with livedo reticularis. Divry–van Bogaert syndrome, with livedo racemosa, seizures, and significant CNS disease, may be related to moyamoya or Sneddon syndrome.
Oxalosis may lead to livedo reticularis from deposition of oxalate crystals in and around blood vessel walls. The characteristic crystals are seen on biopsy. Calciphylaxis, with calcium deposits in vessels and tissue, may cause livedo. Other possible causes of livedo include cryofibrinogenemia, Graves disease (associated with anticardiolipin antibodies), atrial myxoma, tuberculosis (perhaps as a complication of vascular inflammation: vascular-based tuberculid), and syphilis.
Danowski KM, et al: Hydrophilic polymer embolization. J Cutan Pathol 2014; 41: 813.
Dean SM: Livedo reticularis and related disorders. Curr Treat Options Cardiovasc Med 2011; 13: 179.
Gibbs MB, et al: Livedo reticularis. J Am Acad Dermatol 2005; 52: 1009.
Lenert P, et al: ANA(+) ANCA(+) systemic vasculitis associated with the use of minocycline. Clin Rheumatol 2013; 32: 1099.
Quaresma MV, et al: Amantadine-induced livedo reticularis. An Bras Dermatol 2015; 90: 745.
Sangle SR, et al: The prevalence of abnormal pulse wave velocity, pulse contour analysis and ankle-brachial index in patients with livedo reticularis. Rheumatology (Oxford) 2013; 52: 1992.
Tektonidou MG, et al: Antiphospholipid syndrome nephropathy in patients with systemic lupus erythematosus and antiphospholipid antibodies. Arthritis Rheum 2004; 50: 2569.
Tietjen GE, et al: Livedo reticularis and migraine. Headache 2002; 42: 352.
Cholesterol Emboli
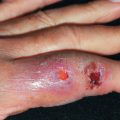
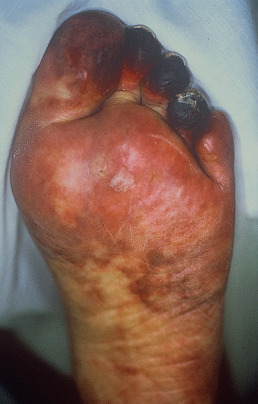
Cholesterol emboli resulting from severe atherosclerotic disease, usually of the abdominal aorta, may cause unilateral or bilateral livedo of the lower extremities. The livedo may not be present with the patient supine and may only be present when the legs are dependent. Patients frequently have concomitant cyanosis (blue toes), purpura, nodules, ulceration, or gangrene ( Fig. 35.4 ). Pain often accompanies the skin lesions. Acute renal failure occurs in up to 75%, and about one third of patients will have characteristic skin lesions. An eosinophilia on complete blood count (CBC) is present in 80% of cases. Older men with severe atherosclerotic disease are at greatest risk. They are often receiving anticoagulant therapy, and many have recently undergone vascular surgery or instrumentation. Slightly more than 1% of left-sided heart catheterizations are complicated by cholesterol emboli. The differential diagnosis includes vasculitis, septic staphylococcal emboli resulting from endocarditis or an infected aneurysm, and polyarteritis nodosa. Cholesterol emboli can involve all organs except the lungs; therefore disease burden can range from mild to overwhelming. Mortality rate can be significant, as high as 90% among those with multisystem involvement, in whom renal failure, bowel infarction, and other devastating complications can occur. Livedo reticularis of recent onset in an elderly person warrants consideration of this diagnosis. Deep biopsy with serial sections may demonstrate the characteristic cholesterol clefts within thrombi. Frozen-section evaluation with polarized microscopy is particularly sensitive. Retinal emboli occur in up to 25% of patients, so funduscopic examination can also aid in diagnosis. Low-dose corticosteroids may be useful for treatment of cholesterol emboli–associated renal insufficiency.
Fukumoto Y, et al: The incidence and risk factors of cholesterol embolization syndrome, a complication of cardiac catheterization. J Am Coll Cardiol 2003; 42: 211.
Jucgla A, et al: Cholesterol embolism. J Am Acad Derm 2006; 55: 786.
Masuda J, et al: Use of corticosteroids in the treatment of cholesterol crystal embolism after cardiac catheterization. Intern Med 2013; 52: 993.
Evaluation of the Patient With Possible Cutaneous Vascular Disorders
In the evaluation of patients who present with livedo, purpura, or ulceration, a broad differential diagnosis must be considered. The diseases considered should include primary pathology of the cutaneous vasculature. In general, these vascular disorders of the skin are divided into two main groups: vasculitis and vasculopathy. Vasculitis includes disorders in which the primary damage in the blood vessels results from inflammatory cells infiltrating and damaging vessel walls. As a consequence of inflammation within vessels, the clotting cascade is triggered, and subsequent thrombosis may be seen in and adjacent to involved vessels. In vasculopathy, the primary process is thrombosis. This is usually caused by a hypercoagulable state. Once thrombosis occurs, inflammatory cells enter the vessel and vessel wall in an attempt to reestablish local circulation. Thus late in a primary thrombotic process, vascular inflammation is seen and can be misinterpreted as “vasculitis.” Emboli can result in a similar histologic picture, because late embolic lesions may also be inflammatory and histologically misleading. All these processes—vasculitis, vasculopathy, and emboli—alter cutaneous blood flow and can be accompanied by livedo. If vessels lose competence, they leak, creating purpura. If vasculitis, vasculopathy, or embolus is severe enough or affects a large enough vessel, the viability of the overlying skin is compromised, and necrosis and ulceration may occur.
Because these entities resemble one another both clinically and histologically, accurate diagnosis is difficult for even the most skilled dermatologist. Careful sampling of early lesions, with large and deep biopsies, if necessary, may be required to find the “primary” vascular pathology. Because vasculitis can be a focal process, step sections may be required to find the diagnostic features. In addition, the diagnosis proposed must be interpreted in the context of other elements of the patient’s medical condition, such as medications, infections, underlying diseases, and involvement of other organ systems besides the skin.
Hirschmann JV, Raugi GJ: Blue (or purple) toe syndrome. J Am Acad Dermatol 2009; 60: 1.
Livedoid Vasculopathy
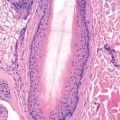
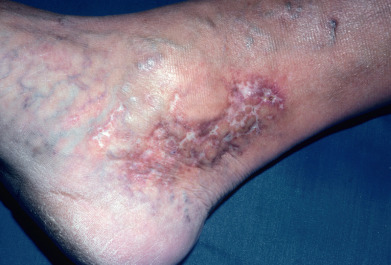
Synonyms for livedoid vasculopathy include livedoid vasculitis, atrophie blanche, segmental hyalinizing vasculitis, livedo reticularis with summer/winter ulceration, and painful purpuric ulcers with reticular pattern of the lower extremities (“PURPLE”). It is a hyalinizing, thrombo-occlusive vascular disease characterized by clotting of medium-sized arterioles. The disorder is chronic, recurrent, and painful. Clinically, purpuric macules and papules cluster around the lower legs, ankles, and dorsal feet. These lesions may develop a hemorrhagic crust, then break down to form irregular, superficial ulcers bordered by violaceous erythema. Over many months, the ulcers heal with porcelain-white, atrophic scars with peripheral telangiectasias, termed atrophie blanche ( Fig. 35.5 ). Other cutaneous findings, such as livedo reticularis, may also be present. About two thirds to three quarters of patients are female; mean age of onset is 45 years. The condition is bilateral in 80% of patients, and ulceration occurs in 70%.
This clinical presentation must be distinguished from other disorders that can cause purpura and ulcers. The differential diagnosis is broad because many conditions can cause livedo reticularis with ulceration of the lower extremities. Atrophie blanche–like lesions are a fairly common end result of ulceration and are therefore not specific for livedoid vasculopathy.
Conditions that mimic livedoid vasculopathy and must be excluded include, most important, the vasculitides—cutaneous small-vessel vasculitis, cryoglobulinemic vasculitis, ANCA-associated vasculitis, and polyarteritis nodosa. Vasculitis involving medium-sized cutaneous vessels can present with ulceration and atrophic, stellate scarring. The presence of other systemic manifestations typical for these conditions should help differentiate them from livedoid vasculopathy. Venous insufficiency, arterial insufficiency, and traumatic ulceration may heal with atrophie blanche and therefore mimic livedoid vasculopathy. Features such as lower extremity edema, hemosiderosis, and venous varicosities may suggest the presence of venous insufficiency, whereas absent pulses, cool extremities, diminished hair growth, and severe pain are typical of arterial insufficiency. A history of trauma should be obtained. A history of characteristic ulcers should be used to distinguish livedoid vasculopathy from other disorders that can lead to atrophic scarring. Dermatoscopic features include central crusted ulcers or ivory white areas with peripheral reticulated pigmentation, and increased vascular structures. Ultimately, in the absence of any contraindication, biopsy should be used to confirm the diagnosis and exclude other causes of ulceration, especially vasculitis.
Biopsy of an affected area must be sufficiently deep to sample medium-sized vessels in the deep dermis or subcutis. Typical findings include hyalinized, thickened dermal blood vessels with fibrin deposition and focal thrombosis. Perivascular hemorrhage and mild perivascular lymphocytic infiltrate can be seen. Notably, no leukocytoclasis or true vasculitis is seen. Results of direct immunofluorescence (DIF) studies are nonspecific. Biopsies of older lesions of atrophie blanche may be most notable for epidermal atrophy and flattening of the rete ridges, as well as recanalization of occluded vessels. In about 15% of patients, an initial biopsy does not reveal diagnostic histology, and a second is required. After two biopsies, diagnostic pathology is found in 98% of patients.
Livedoid vasculopathy is a vasoocclusive condition, a hypercoaguable state with spontaneous thrombosis leading to local hypoxia and skin ulceration. A variety of risk factors for thromboembolism have been identified in association with livedoid vasculopathy. These include genetic and acquired disorders predisposing to thrombosis such as factor V Leiden mutation; protein C or S deficiency; hyperhomocysteinemia, which results in increased clotting; increased plasminogen activator inhibitor (PAI)–1, an important inhibitor of the fibrinolytic system; methylenetetrahydrofolate reductase gene mutation; increased platelet aggregation; low tissue-type plasminogen activator (tPA) levels; enhanced fibrin formation; high levels of lipoprotein A; antithrombin III deficiency; antiphospholipid antibodies; physiologic decrease in levels of protein C and S, as in pregnancy; and other underlying hypercoaguable states (e.g., connective tissue diseases, malignancies).
A review of 45 patients with livedoid vasculopathy included 29 with hypercoaguable workup, 12 of whom (41%) had abnormalities, some multiple. In addition, a number of patients were noted to have connective tissue disease, solid-organ carcinoma, or hematologic malignancy. Livedoid vasculopathy was associated with a comorbid disease or procoagulant state in 58% (26/45). This likely represents an underestimate because exhaustive coagulation screening was not performed in most patients. In a prospective study of 34 patients, 52% (18 patients) screened had laboratory evidence of a coagulopathy, of whom 32% (11) responded to anticoagulant therapy. Some patients diagnosed with “idiopathic” livedoid vasculopathy, in whom no associated abnormality is found, actually have underlying hypercoaguable states discernible only after subsequent, more thorough, workup. As testing for coagulation abnormalities evolves, a greater percentage of livedoid vasculopathy cases will likely be associated with underlying disorders.
Livedoid vasculopathy has been associated with deep venous thrombosis, pulmonary embolism, and cerebrovascular accident (stroke), among other systemic thromboembolic events. Limited data exist to guide management, but in general, treatment of livedoid vasculopathy should be directed at treating the underlying hypercoaguable state, if any. In patients with coagulopathy or personal or family history of thromboembolism, more aggressive therapy may be warranted. A therapeutic ladder for treatment of livedoid vasculopathy begins with local wound care and compression for edema, along with basic measures to decrease risk of thromboembolism (e.g., smoking cessation), followed by the addition of relatively low-risk pharmacologic interventions (e.g., pentoxifylline or aspirin, or vasodilatory agents), before moving on to anticoagulants (e.g., warfarin, low-molecular-weight heparin, rivaroxaban), and other agents with a less favorable risk profile, if needed. Rivaroxaban was studied in a single-arm, open-label study of 28 patients and reduced pain, with minor bleeding in 24% of treated patients and one case of severe menorrhagia. Exceptions to this order might include the introduction of hydroxychloroquine for connective tissue disease, folic acid and vitamin B complex for methylenetetrahydrofolate reductase mutation, danazol or stanozolol for cryofibrinogenemia, and warfarin for antiphospholipid antibody syndrome or a history of systemic thromboembolism. Colchicine has also been used in limited reports. Elevated lipoprotein (a), a risk factor for cardiovascular disease, has been reported in some patients with livedoid vasculopathy, and one study demonstrated danazol improving lipoprotein (a) and healing livedoid vasculopathy–related ulcers. Controlled trials are needed to define better the role of these agents in the treatment of livedoid vasculopathy, as well as the possible role of therapy in preventing systemic thromboembolic complications. Systemic immunosuppression is usually not beneficial for livedoid vasculopathy, because its pathogenesis is thrombo-occlusive, not inflammatory. A dramatic response to high-dose corticosteroids, for example, suggests an alternate diagnosis. IVIGs have been reported as beneficial but are highly viscous and can induce thrombotic events as well.
Alavi A, et al: Livedoid vasculopathy. J Am Acad Dermatol 2013; 69: 1033.
Alavi A, et al: Atrophie blanche. Adv Skin Wound Care 2014; 27: 518.
Callen JP: Livedoid vasculopathy. Arch Dermatol 2006; 142: 1481.
Criado PR, et al: Livedoid vasculopathy and high levels of lipoprotein (a). Dermatol Thera 2015; 28: 248.
Davis MD, Wysokinski WE: Ulcerations caused by livedoid vasculopathy associated with a prothrombotic state. J Am Acad Dermatol 2008; 58: 512.
Deng A, et al: Livedoid vasculopathy associated with plasminogen activator inhibitor-1 promoter homozygosity (4G/4G) treated successfully with tissue plasminogen activator. Arch Dermatol 2006; 142: 1466.
Di Giacomo TB, et al: Frequency of thrombophilia determinant factors in patients with livedoid vasculopathy and treatment with anticoagulant drugs. J Eur Acad Dermatol Venereol 2010; 24: 1340.
Errichetti E, Stinco G: Recalcitrant livedoid vasculopathy associated with hyperhomocysteinaemia responding to folic acid and vitamin B 6 /B 12 supplementation. Acta Derm Venereol 2016; 96: 987.
Gotlib J, et al: Heterozygous prothrombin G20210A gene mutation in a patient with livedoid vasculitis. Arch Dermatol 2003; 139: 1081.
Hairston BR, et al: Treatment of livedoid vasculopathy with low-molecular-weight heparin: report of 2 cases. Arch Dermatol 2003; 139: 987.
Hairston BR, et al: Livedoid vasculopathy. Arch Dermatol 2006; 142: 1413.
Hu SC, et al: Dermoscopic features of livedoid vasculopathy. Medicine (Baltimore) 2017; 96: e6284.
Irani-Hakime NA, et al: Livedoid vasculopathy associated with combined prothrombin G20210A and factor V (Leiden) heterozygosity and MTHFR C677T homozygosity. J Thromb Thrombolysis 2008; 26: 31.
Kim EJ, et al: Pulsed intravenous immunoglobulin therapy in refractory ulcerated livedoid vasculopathy. Dermatol Ther 2015; 28: 287.
Kirsner RS: New hope for patients with livedoid vasculopathy. Lancet Haematol 2016;3(2):e56-7.
Meiss F, et al: Livedoid vasculopathy. Eur J Dermatol 2006; 16: 159.
Monshi B, et al: Efficacy of intravenous immunoglobulins in livedoid vasculopathy. J Am Acad Dermatol 2014; 71: 738.
Rampf J, et al: Methylenetetrahydrofolate-reductase polymorphism associated homocysteinemia in a patient with livedo vasculopathy. Br J Dermatol 2006; 155: 850.
Vasudevan B, et al: Livedoid vasculopathy. Indian J Dermatol Venereol Leprol 2016; 82: 478.
Weishaupt C, et al: Anticoagulation with rivaroxaban for livedoid vasculopathy (RILIVA). Lancet Haematol 2016; 3: e72.
Yong AA, et al: Livedoid vasculopathy and its association with factor V Leiden mutation. Singapore Med J 2012; 53: e258.
Calciphylaxis
Calciphylaxis (calcific uremic arteriolopathy) is an increasingly reported and frequently fatal syndrome that occurs most often in the setting of chronic renal failure but may also occur with normal renal function (nonuremic calciphylaxis). In calciphylaxis, progressive calcification of the media of arterioles leads to vessel injury, intimal fibrosis, and thrombosis, followed by ischemic necrosis of the skin and soft tissue. About 1%–4% of patients on hemodialysis and 4% of patients on peritoneal dialysis develop calciphylaxis. About half of patients are diabetic, and more than half have a body mass index (BMI) greater than 30; every gain in BMI of 1 point over 30 increases the risk for calciphylaxis by 10%. Women outnumber men 3 : 1 to 4 : 1. Other identified risk factors include liver disease, hypoalbuminemia, protein C deficiency, and exposure to warfarin or systemic glucocorticoids. Warfarin in particular is a recognized risk factor, both for uremic and nonuremic calciphylaxis. A strong association between calciphylaxis and thrombophilia has been recently reported in multiple studies, and patients with calciphylaxis should be evaluated for underlying hypercoaguable disorders.
The pathogenesis of calciphylaxis remains poorly understood. Precipitation of calcium phosphate in vessel walls is generally thought to be mediated by elevated serum calcium, phosphate, and parathyroid hormone (PTH) levels, as are seen in chronic renal failure. Indeed, PTH levels are often elevated in affected patients, and the disease can be seen in the setting of primary hyperparathyroidism, as well as the secondary hyperparathyroidism of chronic renal failure. Calcium-phosphate product is greater than 70 in about 20% of calciphylaxis patients. However, a case control study showed no statistical difference in serum calcium, phosphate, PTH, or calcium-phosphate product in patients with calciphylaxis compared with other dialysis patients. Calcium and phosphate are measured at a moment in time, and often calciphylaxis is diagnosed in hospitalized patients who are actively being dialyzed, but who may have missed treatments before admission—thus the measured Ca × P product at time of diagnosis may not be representative of the typical levels. Additionally, experts have suggested that a normal Ca × P product may result from a patient’s inability to maintain those ions in solution, and a higher risk of intravascular calcification. Calcium ingestion, as in the form of calcium-containing phosphate binders, did increase risk. Increasingly, a potential role of vitamin K in the pathogenesis has been suggested, as matrix Gla protein is a vitamin K–dependent inhibitor of extraosseous calcification, and warfarin, which downregulates vitamin K–dependent proteins, is a known risk factor, particularly for nonuremic calciphylaxis.
Calciphylaxis may be best thought of as a disease resulting from exposure of a susceptible host with dysfunctional calcium homeostasis to a particular “challenging” agent or precipitating factor, such as metal salts, fluctuation in renal function, or vascular inflammation. For calcification to occur, vascular smooth muscle cells must transform into osteoblast-like cells. Skin lesions in calciphylaxis exhibit significant upregulation of bone morphogenic protein 2 (BMP-2) and increased expression of inactive uncarboxylated matrix Gla protein (Glu-MGP), osteopontin, fibronectin, laminin, and collagen I, indicating extensive remodeling of the subcutaneous extracellular matrix. Calciphylaxis has repeatedly been seen in patients with Polyneuropathy, organomegaly, endocrinopathy, monoclonal plasmaproliferative disorder, skin changes (POEMS) syndrome, suggesting a possible link to vascular endothelial growth factor (VEGF).
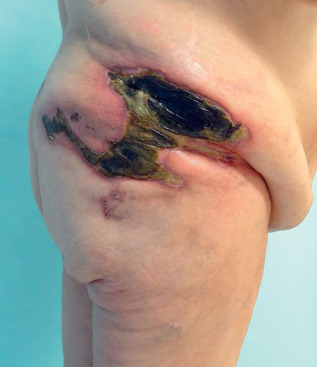
Calciphylaxis begins as fixed livedo reticularis (livedo racemosa), which is frequently firm or hard to the touch and very tender. Areas within the livedo become increasingly violaceous, often with areas of lighter, almost white, blanched skin (due to ischemia), and eventually the area becomes purpuric, bullous, and necrotic. Affected tissue has reduced oxygenation. Lesions affect the legs below the knees in the majority of patients. More proximal lesions and those of the fatty areas of the thighs, buttocks, and abdomen occur in about two thirds ( Fig. 35.6 ). Severe pain is a cardinal feature of calciphylaxis, often requiring narcotic analgesia for control. Ischemic myopathy may occur in severe cases, and muscle pain may precede the appearance of the skin lesions. Penile calciphylaxis is a particularly painful variant. The glans penis develops a deep necrotic ischemic ulceration. Penectomy is often required for pain management. Calciphylaxis of the temporal artery may resemble temporal arteritis.
Necrotic skin lesions are resistant to healing, and infection of open wounds with septicemia is the most common cause of death. The mortality rate of calciphylaxis patients is about 50%–60% at 6–12 months and 80% at 2 years; more proximal lesions portend a worse prognosis, and for patients with both proximal and distal disease, the 1-year mortality rate is 90%. Mortality rate doubles in those with ulcerative lesions. Patients with warfarin-associated nonuremic calciphylaxis appear to have a better prognosis.
Skin biopsy is still the gold standard for diagnosis of calciphylaxis, though opinions differ on whether it should be considered mandatory, as there are concerns that biopsy can induce ulceration and increase the risk for infection. If performed, biopsy should be adjacent to the necrotic area where there is erythema or early purpura, and it should be deep and large enough to identify diagnostic features. This may require an incisional rather than a simple punch biopsy. Vascular calcification is common in all patients with chronic renal failure, so this alone cannot confirm the diagnosis. In addition, there should be evidence of tissue damage (necrosis), extravascular calcification, and thrombosis in the arterioles of the dermis and subcutaneous tissue. Dermal angioplasia is frequently seen, likely due to reactivity to local tissue ischemia. Because the sensitivity of biopsy may be poor, and the clinical presentation often strongly suggests the diagnosis, the true importance of skin biopsy in calciphylaxis is uncertain. Plain x-ray films of affected areas may reveal a characteristic netlike pattern of calcification, though this may not distinguish from atherosclerotic changes. Computed tomography, ultrasound, and even mammography have been used to identify superficial vascular calcifications and may be used to suggest calciphylaxis in an appropriate clinical setting. Nuclear imaging with bone scintigraphy may also demonstrate features suggestive of calciphylaxis, though the sensitivity may vary at different centers.
Much of the treatment for calciphylaxis is directed at altering abnormal calcium metabolism. Because of its high mortality rate, patients are frequently treated with multiple agents at once, making the efficacy of any single agent particularly difficult to determine. Potential exacerbating or triggering agents, such as calcium and iron, should be stopped. In particular, vitamin K antagonists such as warfarin should be stopped, and patients who require anticoagulation should be transitioned to alternate agents. Combination therapy is increasingly favored, and retrospective data appear to support its use. Low-calcium dialysate, non–calcium carbonate phosphate binders, cinacalcet, bisphosphonates, and IV sodium thiosulfate have all been used with some success, with IV sodium thiosulfate now the mainstay of most treatment regimens. Nephrologists should consider transitioning patients on peritoneal dialysis to hemodialysis. Medically treating hyperparathyroidism, if present, is suggested. Parathyroidectomy is best reserved for patients refractory to the previous regimens who have continued marked PTH elevation. One study suggested improved outcomes in patients with stage 5 chronic kidney disease and hyperparathyroidism who underwent parathyroidectomy. Pain control is essential, and patients should be monitored closely for signs of infection. Intralesional sodium thiosulfate, if tolerated (injections are quite painful), has been reported as beneficial in small reports for localized calciphylaxis, and may be an option for patients where IV treatment is not possible, such as nonuremic patients who do not receive dialysis. Beneficial treatment with oral sodium thiosulfate has been reported in a limited number of cases as well. Pentoxifylline has been used anecdotally to improve blood flow and aid in ulcer healing. Other vasodilators, such as alprostadil, have also been reported as beneficial in case reports. Patients with calciphylaxis treated with sodium thiosulfate may have a higher fracture risk, suggesting a potential dual benefit for bisphosphonates, warranting further study. Notably, renal transplantation has resolved calciphylaxis.
Once ulcer or eschar develops, supportive care to ensure no biofilm or superficial bacterial colonization occurs, and topical antibiotics, enzymatic debridement, and careful wound care are all essential to healing any ulcerated areas and preventing secondary infection and sepsis. Hyperbaric oxygen therapy may be a useful adjunctive therapy for ulcer healing. Once ulcerations are present, gentle debridement is associated with healing and increased survival. Becaplermin has been reported to help heal one patient with calciphylaxis ulcers. Surgical debridement has been suggested to improve mortality rate in retrospective studies, with the caveat that patients who were treated surgically tended to have more mild disease and/or fewer comorbidities. There is an actively enrolling clinical trial evaluating vitamin K as a treatment for calciphylaxis, and some suggest repleting low levels if present.
Bhat S, et al: Complete resolution of calciphylaxis after kidney transplantation. Am J Kidney Dis 2013; 62: 132.
Bonchak JG, et al: Calciphylaxis. Int J Dermatol 2016; 55: e275.
Brandenburg VM, et al: Calcific uraemic arteriolopathy (calciphylaxis). Nephrol Dial Transplant 2017; 32: 126.
Brandenburg VM, et al: Lack of evidence does not justify neglect: how can we address unmet medical needs in calciphylaxis? Nephrol Dial Transplant 2016; 31: 1211.
Carter A, et al: Calciphylaxis with evidence of hypercoagulability successfully treated with unfractionated heparin. Clin Exp Dermatol 2016; 41: 275.
Chen TY, et al: Histopathology of calciphylaxis. Am J Dermatopathol 2017; 39: 795.
Dobry AS, et al: Fractures in calciphylaxis patients following intravenous sodium thiosulfate therapy. J Eur Acad Dermatol Venereol 2017; 31: e445.
Jeong HS, Dominguez AR: Calciphylaxis. Am J Med Sci 2016; 351: 217.
Halasz CL, et al: Calciphylaxis. J Am Acad Dermatol 2017; 77: 241.
Hanafusa T, et al: Intractable wounds caused by calcific uremic arteriolopathy treated with bisphosphonates. J Am Acad Dermatol 2007; 57: 1021.
Hayashi M: Calciphylaxis. Clin Exp Nephrol 2013; 17: 498.
Heck D, et al: POEMS syndrome, calciphylaxis, and focal segmental glomerulosclerosis—VEGF as a possible link. BMC Neurol 2014;14: 210.
Kramann R, et al: Novel insights into osteogenesis and matrix remodelling associated with calcific uraemic arteriolopathy. Nephrol Dial Transplant 2013; 28: 856.
McCarthy JT, et al: Survival, risk factors, and effect of treatment in 101 patients with calciphylaxis. Mayo Clin Proc 2016; 91: 1384.
Nigwekar SU, et al: Sodium thiosulfate therapy for calcific uremic arteriolopathy. Clin J Am Soc Nephrol 2013; 8: 1162.
Nigwekar SU, et al: Calciphylaxis. Am J Kidney Dis 2015; 66: 133.
Ning MS, et al: Sodium thiosulfate in the treatment of non-uremic calciphylaxis. J Dermatol 2013; 40: 649.
Ossorio-Garcia L, et al: Multimodal treatment of calciphylaxis with sodium thiosulfate, alrpostadil, and hyperbaric oxygen. Actas Dermosifiliogr 2016; 107: 695.
Paul S, et al: The role of bone scintigraphy in the diagnosis of calciphylaxis. JAMA Dermatol 2017; 153: 101.
Salmhofer H, et al: Multi-modal treatment of calciphylaxis with sodium-thiosulfate, cinacalcet and sevelamer including long-term data. Kidney Blood Press Res 2013; 37: 346.
Schliep S, et al: Successful treatment of calciphylaxis with pamidronate. Eur J Dermatol 2008; 18: 554.
Shetty A, Klein J: Treatment of calciphylaxis. Adv Perit Dial 2016; 32: 51.
Shmidt E, et al: Net-like pattern of calcification on plain soft-tissue radiographs in patients with calciphylaxis. J Am Acad Dermatol 2012; 67: 1296.
Strazzula L, et al: Intralesional sodium thiosulfate for the treatment of calciphylaxis. JAMA Dermatol 2013; 149: 946.
Tian F, et al: The cutaneous expression of vitamin K-dependent and other osteogenic proteins in calciphylaxis. J Am Acad Dermatol 2016; 75: 840.
Twu O, et al: Use of becaplermin for nondiabetic ulcers. Dermatol Ther 2016; 29: 104.
Vedvyas C, et al: Calciphylaxis. J Am Acad Dermatol 2012; 67: e253.
Weenig RH, et al: Calciphylaxis. J Am Acad Dermatol 2007; 56: 569.
Yu WY, et al: Warfarin-associated nonuremic calciphylaxis. JAMA Dermatol 2017; 153: 309.
Marshall-White Syndrome and Bier Spots
Bier spots are pale, irregularly shaped macules about 10 mm in size, usually found on the upper and lower extremities of young adults. The spots are a type of vascular mottling that can be elicited by placing the limbs in a dependent position; they resolve when the limbs are raised and disappear when the surrounding skin is blanched. They likely represent areas of localized vasoconstriction surrounded by relative vasodilation. Although primarily idiopathic, asymptomatic, and transient, there are case reports of Bier spots in association with such disorders as cryoglobulinemia and scleroderma renal crisis and with pregnancy. Awareness of the condition can prevent misdiagnosis of a pigmentary disorder.
Fan YM, et al: Bier spots. J Am Acad Dermatol 2009; 61: e11.
Mahajan VK, et al: Bier spots. Indian Dermatol Online J 2015; 6: 128.
Purpura
Purpura is the term used to describe extravasation of blood into the skin or mucous membranes. It presents as distinctive, brownish red or purplish macules a few millimeters to many centimeters in diameter. Several terms are used to describe various clinical manifestations of purpura.
Petechiae are superficial, pinhead-sized (<3 mm), round, hemorrhagic macules, bright red at first, then brownish or rust colored. They are most often seen in dependent areas, occur in crops, regress over days, and usually imply a disorder of platelets rather than of coagulation factors, which typically give rise to ecchymoses or hematomas rather than petechiae.
Ecchymoses are commonly known as bruises. These extravasations signify a deeper, more extensive interstitial hemorrhage that forms a flat, irregularly shaped, blue-purple patch. Such patches gradually turn yellow and finally fade away.
Vibices (singular, vibex) are linear purpuric lesions.
Hematoma designates a pool-like collection of extravasated blood in a dead space in tissue that, if of sufficient size, produces swelling that fluctuates on palpation. Hematomas are usually walled off by tissue planes.
Pathogenesis
Purpura may result from hypercoagulable and hypocoagulable states, vascular dysfunction, idiopathic thrombocytopenic purpura, TTP, disseminated intravascular coagulation (DIC), drug-induced thrombocytopenia, bone marrow failure, congenital or inherited platelet function defects, acquired platelet function defects (aspirin, renal or hepatic disease, gammopathy), and thrombocytosis secondary to myeloproliferable diseases. Most of these disorders produce findings of nonpalpable purpura. Ecchymosis predominates in procoagulant defects, such as hemophilia, pharmacologic anticoagulation, vitamin K deficiency, and advanced hepatic disease resulting in impaired synthesis of clotting factors. There is often a component of trauma. Increased ecchymosis can be the result of poor dermal support of blood vessels, most often localized to the area of trauma, and may result from actinic (senile) purpura, topical or systemic corticosteroid therapy, scurvy, systemic amyloidosis, Ehlers-Danlos syndrome, or pseudoxanthoma elasticum.
Prothrombotic disorders form characteristic “retiform” purpura or purpura associated with livedo reticularis. These include disorders in which fibrin, cryoglobulin, or other material occludes vessels. Representative causes include monoclonal cryoglobulinemia, cryofibrinogenemia, DIC, purpura fulminans, protein C or S deficiency, warfarin-induced necrosis, heparin necrosis, cholesterol emboli, oxalate crystal occlusion, and antiphospholipid syndrome.
Evaluation
A history and physical examination are often sufficient to evaluate for purpura. A family history of bleeding or thrombotic disorders, duration of symptoms, use of drugs and medications that might affect platelet function and coagulation, and review of medical conditions that may result in altered coagulation should be documented. Physical examination should stress the size, type, and distribution of purpura; a search for telangiectasias; a joint examination; and an evaluation of skin elasticity, unusual scars, and unusual body habitus. Correlation of purpura morphology with pathogenesis allows for a more focused approach.
A CBC and differential can be used to assess for microangiopathic anemia, screen for myeloproliferative disorders, and assess the number and morphology of platelets. A bleeding time is the preferred method of assessing platelet function. The partial thromboplastin time (PTT) and prothrombin time (PT) are tests to evaluate abnormal coagulation states.
Thrombocytopenic Purpura
Thrombocytopenic purpura may be classified into two large categories: states resulting from accelerated platelet destruction and states resulting from deficient platelet production. Accelerated platelet destruction may be immunologic or nonimmunologic. The former may be caused by antibodies (autoimmune or drug-induced thrombocytopenia), isoantibodies (congenital or posttransfusion), immune complex disease, or other immunologic processes, such as erythroblastosis fetalis, neonatal lupus, scleroderma, other connective tissue diseases, or acquired immunodeficiency syndrome (AIDS). The group of thrombocytopenias with accelerated platelet destruction also includes TTP and DIC. Deficient platelet production may be related to diseases such as aplastic anemia and leukemia.
Immune Thrombocytopenic Purpura (Immune Thrombocytopenia)
Immune thrombocytopenic purpura (ITP) was also known as “idiopathic” thrombocytopenic purpura or Werlhof disease. It is an autoimmune disease characterized by an isolated thrombocytopenia (platelet count <100,000). The causative antibodies are directed at molecules on the platelet surface, leading to their premature sequestration and destruction, primarily in the spleen. ITP is called primary in the absence of another cause, or secondary if there is a causal association, such as “secondary ITP (SLE-associated).” Bleeding symptoms are minimal or absent in a large proportion of cases. Cutaneous manifestations can include an acute or gradual onset of petechiae or ecchymoses on the skin and mucous membranes, especially in the mouth. Epistaxis, conjunctival hemorrhages, hemorrhagic bullae in the mouth ( Fig. 35.7 ), and gingival bleeding may occur. Melena and hematemesis are also present, as well as menorrhagia, which may be the first sign of the disease in young women. Chronic leg ulcers occasionally develop.
Bleeding can occur when the platelet count is less than 50,000/mm 3 . Posttraumatic hemorrhage, spontaneous hemorrhage, and petechiae may appear. The risk of serious hemorrhage is greatly increased at levels below 10,000/mm 3 . The most serious complication is intracranial hemorrhage. ITP may be fatal, but most mortality in adults results from treatment complications. Bleeding time is usually prolonged and coagulation time normal, whereas clot retraction time is abnormal and capillary fragility increased. Increased numbers of megakaryocytes are found in the bone marrow.
The age of onset determines the clinical manifestations and course. In children, onset is often acute and follows a viral illness in 50%–60% of patients. Parvovirus B19 is frequently complicated by thrombocytopenia, which may be ITP or simply a consequence of reduced bone marrow production of platelets. The average lag between purpura and the preceding infection is usually 2 weeks (range 1–4 weeks). Most of these cases resolve spontaneously. Because children are at much less risk of developing serious hemorrhagic complications, a more conservative management approach may be taken. A few patients will develop chronic thrombocytopenia, and deaths, usually from cerebral hemorrhage, have been reported. In a series of 332 children with ITP, 58 (17%) had episodes of major hemorrhage. One death resulted from sepsis. In another series of 427 cases, 323 (72%) had mild to benign disease. About 85% of children who undergo splenectomy experience remission. More than half of the remaining patients spontaneously remit within 15 years.
The chronic form of ITP occurs most often in adults, is persistent, and has a female/male ratio of 2 : 1 to 4 : 1. Secondary ITP is more common in adults. Human immunodeficiency virus (HIV) infection, hepatitis C, and autoimmune disease are the most common associated disorders. Treatment of associated disease may lead to improvement of the thrombocytopenia. Breast cancer has been associated with ITP, with a parallel course in one third of cases. Other malignancies have also been associated with ITP. Helicobacter pylori infection as a cause of ITP is controversial, but testing for H. pylori antibodies and treatment for infection carry limited toxicity and thus could be considered.
In elderly patients, ITP is more difficult to manage. Patients more frequently have major bleeding complications, more complications from immunosuppressive agents, especially corticosteroids, and more complications from splenectomy. Corticosteroids have a particularly low response rate in elderly ITP patients. Danazol has demonstrated reasonable safety and efficacy in the elderly population.
The differential diagnosis of ITP includes drug-induced thrombocytopenia, myelodysplasia, TTP, and congenital/hereditary thrombocytopenia. The goal of treatment for ITP is to raise the platelet count above 20,000–30,000 and to stop all bleeding symptoms. Platelet transfusions are indicated if there is significant bleeding or if the platelet count is dangerously low. If the platelet count is greater than 20,000–30,000, the patient may be closely monitored. The treatment of ITP has changed with the availability of new approaches. Initial treatment is a short course of high-dose corticosteroids, either for 1–2 weeks or as monthly pulses. IVIG or anti-(Rh)D, also known as IV Rh immune globulin (IG), may be given with this treatment. Platelet survival is increased if transfused immediately after immunoglobulin infusion. If the patient relapses or has persistent symptoms, systemic corticosteroids are given with rituximab, anti-(Rh)D, IVIG, or a thrombopoietin agonist such as eltrombopag or romiplostim. Splenectomy can be considered a second-line treatment, although age over 60 makes this treatment less desirable. Danazol can be added as a second-line agent, especially in the elderly patient. When second-line treatments have failed in patients with chronic and persistent or worsening disease, immunosuppression with mycophenolate mofetil (MMF), azathioprine, cyclosporine, vincristine, lymphoma-type chemotherapeutic regimens, and even autologous transplantation could be considered.
Aktepe OC, et al: Human parvovirus B19 associated with idiopathic thrombocytopenic purpura. Pediatr Hematol Oncol 2004; 21: 421.
Aledort LM, et al: Prospective screening of 205 patients with ITP, including diagnosis, serological markers, and the relationship between platelet counts, endogenous thrombopoietin, and circulating antithrombopoietin antibodies. Am J Hematol 2004; 76: 205.
Cines DB, Blanchette VS: Immune thrombocytopenic purpura. N Engl J Med 2002; 346: 995.
Daou S, et al: Idiopathic thrombocytopenic purpura in elderly patients. Eur J Intern Med 2008; 19: 447.
George JN: Definition, diagnosis and treatment of immune thrombocytopenic purpura. Haematologica 2009; 94: 759.
Godeau B: Immune thrombocytopenic purpura. Presse Med 2014; 43: e47.
Kuter DJ, et al: Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura. Lancet 2008; 371: 395.
Noonavath RN, et al: Helicobacter pylori eradication in patients with chronic immune thrombocytopenic purpura. World J Gastroenterol 2014; 20: 6918.
Ozkan MC, et al: Immune thrombocytopenic purpura. Curr Med Chem 2015; 22: 1956.
Rodeghiero F, et al: Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children. Blood 2009; 113: 2386.
Drug-Induced Thrombocytopenia
Thrombocytopenic purpura resulting from drug-induced antiplatelet antibodies may be caused by agents such as heparin, sulfonamides (antibiotics and hydrochlorothiazide), digoxin, quinine, quinidine, chlorothiazide, penicillin, cephalosporins, minocycline, acetaminophen, nonsteroidal antiinflammatory drugs (NSAIDs), statins, fluconazole, protease inhibitors, H2 blockers, antiplatelet agents, rifampin, and lidocaine. More broadly, drug-induced immune thrombocytopenia overall can be caused by over 100 drugs, particularly carbamazepine, ibuprofen, quinidine, quinine, oxaliplatin, rifampin, sulfamethoxazole, trimethoprim, and vancomycin. Chemotherapeutic agents, including checkpoint inhibitors, can frequently cause thrombocytopenia.
Heparin-induced thrombocytopenia (HIT) is associated with life-threatening arterial and venous thrombosis and, to a lesser extent, hemorrhagic complications. The platelet count usually begins to fall 4–14 days after starting heparin, more frequently in a patient with prior exposure to the medication. Platelet counts drop to about 50% of their pre-HIT level, usually with a nadir of about 50,000, and rarely 10,000. HIT is mediated by an antibody to the platelet factor 4 (PF4)–heparin complex. The antibody cross-links FcγRII receptors on the platelet surface, resulting in platelet activation, aggregation, and simultaneous activation of blood-coagulation pathways. Tests for HIT antibodies include immunoassays such as enzyme-linked immunosorbent assay (ELISA) and functional tests.
Treatment for drug-induced thrombocytopenia consists of removal of the offending agent. All forms of heparin, including heparin flushes, should be discontinued. Because the HIT antibody continues to activate platelets after heparin cessation, patients with HIT have a persistently high risk of thrombosis and an ongoing need for anticoagulation. A nonheparin anticoagulant (e.g., argatroban, bivalirudin, fondaparinux) should be begun immediately unless there is a strong contraindication to anticoagulation, such as active bleeding or high bleeding risk. Warfarin can cause microthrombosis in these patients, so it should be avoided until after the patient is stably anticoagulated with another agent and the platelet count has recovered to 150,000 or higher. The total duration of anticoagulation should be at least 2–3 months in the absence of a thrombotic event and 3–6 months if thrombosis has occurred.
Chong BH, Isaacs A: Heparin-induced thrombocytopenia. Thromb Haemost 2009; 101: 279.
Curtis BR: Drug-induced immune thrombocytopenia. Immunohematology 2014; 30: 55.
Goldfarb MJ, Blostein MD: Fondaparinux in acute heparin-induced thrombocytopenia. J Thromb Haemost 2011; 9: 2501.
Linkins LA, et al: Treatment and prevention of heparin-induced thrombocytopenia. Chest 2012; 141: e495S.
Reese JA, et al: Identifying drugs that cause acute thrombocytopenia. Blood 2010; 116: 2127.
Shantsila E, et al: Heparin-induced thrombocytopenia. Chest 2009; 135: 1652.
Thrombotic Microangiopathy
The diagnosis of a thrombotic microangiopathy is made in the presence of a microangiopathic hemolytic anemia and thrombocytopenia, in the absence of another plausible explanation. TTP and hemolytic uremic syndrome are the two major diseases in this group. Certain drugs, such as cyclosporine, quinine, ticlopidine, clopidogrel, mitomycin C, docetaxel, trastuzumab, and bleomycin, have been associated with a thrombotic microangiopathy.
Thrombotic Thrombocytopenic Purpura
Classically, TTP consists of the pentad of microangiopathic hemolytic anemia, thrombocytopenic purpura, neurologic abnormalities, fever, and renal disease. However, only the minority of patients (20%–30%) present classically; many patients do not have renal disease, and CNS findings are not required for the diagnosis. The diagnosis of TTP requires only a Coombs-negative microangiopathic hemolytic anemia and thrombocytopenia with platelet aggregation in the microvasculature. Most patients will develop neurologic findings. Fever is present in 75%. Multiple ecchymoses and retiform purpura may be found on the skin. The presence of schistocytes on a blood smear is the morphologic hallmark of the disease, and a schistocyte count greater than 1% in the absence of other known causes of thrombotic microangiopathy strongly suggests a diagnosis of TTP. Tests may show a decreased hematocrit and decreased platelets, an elevated lactate dehydrogenase level, and elevated indirect bilirubin. A delay in diagnosis may lead to a mortality rate as high as 90%. For this reason, the presence of microangiopathic hemolytic anemia and thrombocytopenia in the absence of an obvious cause (e.g., DIC) is justification enough to begin empiric therapy. Biopsies demonstrate hemorrhage and fibrin occlusion of vessels. Inflammation is absent. Studies of plasma samples from patients with active TTP show the presence of unusually large von Willebrand factor (vWF) multimers. The underlying cause of TTP is a congenital or acquired deficiency of the vWF-cleaving protease, ADAMTS13. vWF is secreted by the endothelial cell in long multimers, which should be cleaved into monomers by ADAMTS13 and released into the circulation. Instead, multimers circulate and extend from the surface of the endothelial cells in the microvasculature. Platelets adhere to these multimers and the surface of the endothelial cell, leading to microvascular thrombosis.
The two forms of TTP are idiopathic and congenital (Upshaw-Schulman syndrome). Congenital TTP is less common (<10% of cases) and usually presents in infancy or childhood with jaundice and thrombocytopenia. Some patients with a congenital deficiency of ADAMTS13 do not present until adulthood or may even remain asymptomatic. The course is frequently relapsing TTP at regular intervals. Idiopathic TTP is a rare disease, about 10 cases per 1 million population per year. Women represent 70% of cases, and those of African descent have a ninefold greater risk of developing idiopathic TTP. Idiopathic TTP is caused by an autoantibody directed against ADAMTS13 that can be detected in up to 85% of cases. Neurologic symptoms are the most frequent presentation, ranging from confusion to seizures to coma.
Plasma exchange with fresh frozen plasma is the treatment of choice for TTP. Before it was instituted, 80% of these patients died; now, 80% survive. Plasma exchange clears the vWF multimers, reduces the autoantibody, and replenishes the inhibited ADAMTS13. If plasma exchange is not immediately available, simple plasma infusion can be performed until it can be instituted. Plasma exchange is usually continued daily until clinical symptoms improve and the platelet count is above 150,000, generally about 5–10 total exchanges. In conjunction with plasma exchange, glucocorticoids may be given, although their success is variable. Platelet transfusions should be avoided except in the setting of life-threatening (i.e., CNS) bleeding. Cyclosporine, cyclophosphamide, and rituximab have been used in refractory cases, with promising results. Splenectomy can also be considered. Congenital TTP is usually much easier to treat, with only small amounts of normal plasma infusion required to provide the missing ADAMTS13 and stop the clotting.
Hemolytic Uremic Syndrome
Hemolytic uremic syndrome (HUS) has many similarities to TTP but is now considered a distinct entity, both clinically and pathogenically. HUS is usually a disease of childhood. Patients have a microangiopathic hemolytic anemia, often after a diarrheal illness caused by Shiga toxin (Stx)–producing Escherichia coli. The annual incidence is 6.1 cases per 100,000 population. It is the most common cause of acute renal injury requiring transplantation in children age 1–5. Streptococcus pneumoniae infection can also precipitate HUS. Cases of HUS not following such infections are called “atypical HUS.”
Fever is usually absent in HUS patients. Renal insufficiency occurs in all patients and is the hallmark of HUS. Neurologic disease can occur but affects less than half of patients. Skin involvement is unusual but may take the form of retiform purpura and petechiae.
The pathogenic mechanism of typical HUS is endothelial damage caused by the bacterial toxin and subsequent complement activation on the endothelial surface. The affected vessels are thickened, endothelial cells are detached, and the vascular lumen is narrowed and occluded by platelet thrombi. The renal vessels are at particular risk, because the subendothelial membrane is exposed and vulnerable to complement-mediated damage. In atypical and familial HUS, similar complement activation via the alternative pathway (through C3b) occurs on endothelial surfaces, leading to endothelial damage and intravascular thrombosis.
In atypical and familial HUS, mutations in the alternative complement cascade have been identified. Complement factor H (CFH) mutations are most common, and many patients are heterozygotes. The abnormal CFH complexes with the normal CFH, inactivating it. CFH is the major downregulator of the alternative complement cascade as it degrades C3b. Loss of CFH activity allows for unopposed C3b activity and complement activation. Other mutations are in complement factor I (CFI), which cleaves c3B and C4b. Mutations in membrane cofactor protein (MCP), a cofactor for CFI, in C3 itself, in complement factor B (component of C3b), and in thrombomodulin can also cause atypical HUS. The type of mutation determines the clinical course of atypical HUS, with CFH, CFI, CFB, and thrombomodulin mutations having rates of death or end-stage renal disease of more than 50%. HUS recurs in more than three quarters of patients with CFH and CFI mutations. Some patients are compound heterozygotes with mutations in two of the genes previously noted. About 6% of patients have an autoantibody to CFH and have “autoimmune HUS.”
Although atypical or familial HUS is a disorder caused by a genetic deficiency in most cases, onset may not occur until middle age. About 67% of atypical HUS occurs during childhood, with almost all patients with anti-CFH antibodies diagnosed before age 16. Oral contraceptive (OC) use may trigger HUS in 8% of patients with CFH and 20% of patients with CFI mutations.
The treatment of HUS is primarily supportive, including management of renal failure. Plasma exchange may be used but unfortunately does not have the same degree of benefit in HUS as in TTP. Eculizumab, a humanized monoclonal antibody to C5, is effective for the treatment of complement-mediated HUS caused by CFH and CFI mutations and is the first treatment approved by the U.S. Food and Drug Administration (FDA) for atypical HUS. Some data suggest eculizumab may also be beneficial for Stx-associated HUS, but this remains controversial. The cost of the medication, about $400,000 (U.S.) per year, may be prohibitive.
Corticosteroids, azathioprine, vincristine, MMF, and rituximab may be used in atypical HUS. The role of kidney transplantation in atypical HUS is unclear because of the high rate of recurrence and loss of the graft. The likelihood for a successful outcome after transplantation depends on mutation type.
Ardissino G, et al: Skin involvement in atypical hemolytic uremic syndrome. Am J Kidney Dis 2014; 63: 652.
Azoulay E, et al: Expert statements on the standard of care in critically ill adult patients with atypical hemolytic uremic syndrome. Chest 2017; 152: 424.
Beloncle F, et al: Splenectomy and/or cyclophosphamide as salvage therapies in thrombotic thrombocytopenic purpura. Transfusion 2012; 52: 2436.
Benhamou Y, et al: Development and validation of a predictive model for death in acquired severe ADAMTS13 deficiency–associated idiopathic thrombotic thrombocytopenic purpura. Haematologica 2012; 97: 1181.
Bouw MC, et al: Thrombotic thrombocytopenic purpura in childhood. Pediatr Blood Cancer 2009; 53: 537.
Burns ER, et al: Morphologic diagnosis of thrombotic thrombocytopenic purpura. Am J Hematol 2004; 75: 18.
Cataland SR, Wu HM: How I treat: the clinical differentiation and initial treatment of adult patients with atypical hemolytic uremic syndrome. Blood 2014; 123: 2478.
Delmas Y, et al: Outbreak of Escherichia coli O104:H4 haemolytic uraemic syndrome in France. Nephrol Dial Transplant 2014; 29: 565.
Feys HB, et al: ADAMTS13 in health and disease. Acta Haematol 2009; 121: 183.
Freedman SB, et al: Shiga toxin-producing Escherichia coli infection, antibiotics, and risk of developing hemolytic uremic syndrome. Clin Infect Dis 2016; 62: 1251.
Froissart A, et al: Efficacy and safety of first-line rituximab in severe, acquired thrombotic thrombocytopenic purpura with a suboptimal response to plasma exchange. Crit Care Med 2012; 40: 104.
George JN: How I treat patients with thrombotic thrombocytopenic purpura. Blood 2010; 116: 4060.
Kielstein JT, et al: Best supportive care and therapeutic plasma exchange with or without eculizumab in Shiga-toxin-producing E. coli O104:H4 induced haemolytic-uraemic syndrome. Nephrol Dial Transplant 2012; 27: 3807.
Legendre CM, et al: Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med 2013; 368: 2169.
Lotta LA, et al: ADAMTS13 mutations and polymorphisms in congenital thrombotic thrombocytopenic purpura. Hum Mutat 2010; 31: 11.
Marques MB: Thrombotic thrombocytopenic purpura and heparin-induced thrombocytopenia. Clin Lab Med 2009; 29: 321.
Menne J, et al: Validation of treatment strategies for enterohaemorrhagic Escherichia coli O104:H4 induced haemolytic uraemic syndrome: case-control study. BMJ 2012; 345: e4565.
Nayer A, Asif A: Atypical hemolytic-uremic syndrome. Am J Ther 2016; 23: e151.
Scully M, et al: Guidelines on the diagnosis and management of thrombotic thrombocytopenic purpura and other thrombotic microangiopathies. Br J Haematol 2012; 158: 323.
Siau K, Varughese M: Thrombotic microangiopathy following docetaxel and trastuzumab chemotherapy. Med Oncol 2010; 27: 1057.
Tsai HM: Thrombotic thrombocytopenic purpura and the atypical hemolytic-uremic syndrome: an update. Hematol Oncol Clin North Am 2013; 27: 565.
Yilmaz M, et al: Cyclosporin A therapy on idiopathic thrombotic thrombocytopenic purpura in the relapse setting. Transfusion 2013; 53: 1586.
Nonthrombocytopenic Purpura (Dysproteinemic Purpura)
Cryoglobulinemia and Cryofibrinogenemia
The term cryoglobulinemia refers to the presence in the serum of proteins that precipitate at temperatures below 37°C and redissolve on rewarming. These tend to be chronic conditions, unless the underlying disease process is treated. Abnormal serum proteins behaving as cryoglobulins and cryofibrinogens may be IgG, IgM, or both. Type I cryoglobulinemia results from monoclonal immunoglobulins, usually IgM and less frequently IgG, IgA, or light chains caused by an underlying lymphoproliferative disorder, usually multiple myeloma or macroglobulinemia. Type II cryoglobulinemia results from monoclonal IgM (rarely IgG and IgA) immunoglobulins, which have rheumatoid factor (RF)–like activity and form complexes to the constant, fragment crystallizable (Fc) region of polyclonal IgG. This can occur in many connective tissue diseases and may also be caused by the B-cell proliferation seen in hepatitis C virus (HCV) infection. Type III cryoglobulinemia (mixed cryoglobulinemia), in which the cryoglobulins are polyclonal and of various classes, is associated with HCV infection in more than 90% of cases, but may be seen with connective tissue disorders or other infections, such as osteomyelitis or endocarditis. Together, types II and III cryoglobulinemia constitute 80% of cases.
Purpura is most likely to occur on exposed surfaces after cold exposure. This may occur in the summer as well as the winter, possibly due to indoor air conditioning, and has been reported in the intensive care unit in acutely febrile patients treated with cooling packs and ice. It may be of sudden onset and may clear rapidly once the patient is kept warm. Marked brown hyperpigmentation of the dorsal feet, at times in a livedoid pattern, may suggest this diagnosis. Cryoglobulinemia can be the cause of chronic leg ulcers. An unusual clinical presentation of type I cryoglobulinemia in association with multiple myeloma is follicular hyperkeratosis of the central face, especially the nose.
Systemic complications in type I cryoglobulinemia relate primarily to hyperviscosity and thrombosis. Manifestations of type II and III disease are multisystem and similar to those of other types of small- to medium-vessel vasculitis, including arthralgias and myalgias, glomerulonephritis, interstitial pulmonary infiltrates, and neuropathy.
In monoclonal disease, the biopsy reveals amorphous, jelly-like, eosinophilic material in the vessel lumen. In types II and III cryoglobulinemia, a skin biopsy reveals classic leukocytoclastic vasculitis ( Fig. 35.8 ) and, less often, features of polyarteritis nodosa.
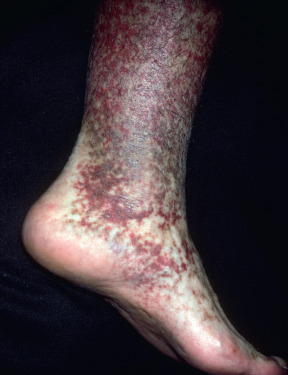
Asymptomatic disease need not be treated. Most patients should be advised to maintain body warmth, both with layers over the core and with appropriate gear to protect the extremities. With symptomatic disease, the aim of therapy is to treat, eliminate, or control the underlying condition and to suppress the associated immune response. For type I cryoglobulinemia, this means addressing the associated myeloproliferative disorder, and for cryoglobulinemia associated with HCV, that means antiviral therapy. Reduction of the HCV viral load is the long-term solution in HCV-associated cases and can result in disappearance of the cryoglobulins. Improved treatment options for HCV are increasingly available, and direct antiviral therapy has led to improvement in HCV-associated cryoglobulinemia. The overall treatment approach varies significantly based on the underlying disease. Systemic corticosteroids, colchicine, thalidomide, immunosuppressants such as cyclophosphamide or azathioprine, and IVIG have all been used. Rituximab is increasingly reported as beneficial, even in cases of infection-associated cryoglobulinemia, and may be necessary to control cryoglobulinemia-related manifestations before or concurrent with antiviral therapy. Plasmapheresis is indicated for severe or refractory disease. Simple plasma exchange can be helpful, but cryofiltration apheresis is the best method to remove cryoproteins in the treatment of cryoprecipitate-induced diseases. Often plasmapheresis or exchange is combined with another therapy to prevent rapid reappearance of the cryoglobulin.
Compared with cryoglobulinemia, cryofibrinogenemia is less often symptomatic and generally more readily treatable. Patients most often present with purpura, skin necrosis, and arthralgias; ulceration and gangrene can result. The precipitating cryofibrinogen is a cold-insoluble complex of fibrin, fibrinogen, and fibrin split products with albumin, cold-insoluble globulin, factor VIII, and plasma proteins. Associated collagen vascular disorders, infections, and malignancies are significantly more common in patients with both cryofibrinogens and cryoglobulins than in those with isolated cryofibrinogenemia. Cryofibrinogen has been associated with calciphylaxis in the setting of renal disease and livedoid vasculopathy when accompanied by other prothrombotic risks. Familial primary cryofibrinogenemia manifests as painful purpura, with slow-healing ulcerations and edema of both feet during the winter months. Therapy beyond cold avoidance is with aspirin, corticosteroids, or stanazol for moderate disease. Immunosuppressive and antithrombotic therapies are given for severe disease. Response rates are high, but relapses are common.
Cacoub P, et al: Cryoglobulinemia vasculitis. Am J Med 2015; 128: 950.
Colantuono S, et al: Efficacy and safety of long-term treatment with low-dose rituximab for relapsing mixed cryoglobulinemia vasculitis. Clin Rheumatol 2017; 36: 617.
Da Silva Fucuta Pereira P, et al: Long-term efficacy of rituximab in hepatitis C virus–associated cryoglobulinemia. Rheumatol Int 2010; 30: 1515.
De Rosa FG, et al: Observations on cryoglobulin testing. I. J Rheumatol 2009; 36: 1953.
De Rosa FG, et al: Observations on cryoglobulin testing. II. J Rheumatol 2009; 36: 1956.
Emery JS, et al: Efficacy and safety of direct acting antivirals for the treatment of mixed cryoglobulinemia. Am J Gastroenterol 2017; 112: 1298.
Ferri C, et al: Mixed cryoglobulinemia. Semin Arthritis Rheum 2004; 33: 355.
Ghetie D, et al: Cold hard facts of cryoglobulinemia. Rheum Dis Clin North Am 2015; 41: 93.
Harati A, et al: Skin disorders in association with monoclonal gammopathies. Eur J Med Res 2005; 10: 93.
Mazzaro C, et al: Efficacy and safety of peginterferon alfa-2b plus ribavirin for HCV-positive mixed cryoglobulinemia. Clin Exp Rheumatol 2011; 29: 933.
Michaud M, Pourrat J: Cryofibrinogenemia. J Clin Rheumatol 2013; 19: 142.
Molinero C, et al: Cryoglobulinemia precipitated by targeted temperature management. Intensive Care Med 2015; 41: 1355.
Muchtar E, et al: How I treat cryoglobulinemia. Blood 2017; 129: 289.
Ramos-Casals M, et al: The cryoglobulinaemias. Lancet 2012; 379: 348.
Roccatello D, et al: Improved (4 plus 2) rituximab protocol for severe cases of mixed cryoglobulinemia. Am J Nephrol 2016; 43: 251.
Saadoun D, et al: Efficacy and safety of sofosbuvir plus daclatasvir for treatment of HCV-associated cryoglobulinemia vasculitis. Gastroenterology 2017; 153: 49.
Sidana S, et al: Clinical presentation and outcomes of patients with type 1 monoclonal cryoglobulinemia. Am J Hematol 2017; 92: 668.
Sinha D, et al: Cryofiltration in the treatment of cryoglobulinemia and HLA antibody-incompatible transplantation. Ther Apher Dial 2012; 16: 91.
Terrier B, et al: The spectrum of type I cryoglobulinemia vasculitis. Medicine (Baltimore) 2013; 92: 61.
Visentini M, et al: Efficacy of low-dose rituximab for the treatment of mixed cryoglobulinemia vasculitis. Autoimmune Rev 2015; 14: 889.
Yang CH, et al: Long-term plasmapheresis in conjunction with thalidomide and dexamethasone for the treatment of cutaneous ulcers and neovascular glaucoma in recalcitrant type I cryoglobulinemia. JAMA Dermatol 2014; 150: 426.
Waldenström Hyperglobulinemic Purpura (Purpura Hyperglobulinemica)
Waldenström hyperglobulinemic purpura presents with episodic showers of petechiae that may burn or sting, occurring mainly on the lower extremities. The dorsa of the feet are intensely involved, and the petechiae diminish on the ascending parts of the feet ( Fig. 35.9 ). A diffuse “peppery” distribution is typically noted, resembling Schamberg disease. The petechiae may be induced or aggravated by prolonged standing or walking or by wearing constrictive garters or stockings.
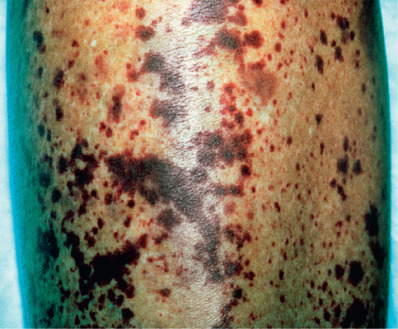
Serum protein electrophoresis demonstrates a broad-based peak (polyclonal hypergammaglobulinemia). The bulk of the protein increase is IgG, although occasionally, increased amounts of IgA are also found. IgM is usually normal or decreased. RF in varying amounts is present in almost all patients. Antithyroglobulins, increased erythrocyte sedimentation rate (ESR), leukopenia, antinuclear factors, and proteinuria may be found. Almost 80% of patients with hypergammaglobulinemic purpura of Waldenström have antibodies to Ro/SSA.
Hyperglobulinemic purpura occurs most frequently in women between ages 18 and 40 and is often seen with Sjögren syndrome and rheumatoid arthritis. Adverse fetal outcomes in these women may be associated with the presence of SSA or SSB autoantibodies. Hyperglobulinemic purpura may also be a primary, chronic, benign illness. When it is associated with hepatitis C, it has a predilection for men and has manifestations that usually last longer than those associated with Sjögren syndrome.
In about one third of patients, leukocytoclastic vasculitis is present. These patients have a higher prevalence of articular involvement, peripheral neuropathy, Raynaud phenomenon, renal involvement, ANA, RF, and anti-Ro/SSA antibodies. The course of the disease is essentially benign but chronic. Rare deaths are related to associated cryoglobulin disease. Hyperglobulinemic purpura may be a manifestation or harbinger of connective tissue or hematopoietic diseases, and rarely, progression to myeloma has been reported.
Patients often improve with support stockings. Corticosteroids should be reserved for severe disease. Indomethacin and hydroxychloroquine may be of value in the treatment of milder disease, especially in patients who have connective tissue disease or are SSA/B (Ro/La) positive. Aspirin and colchicine have been used with some success.
Waldenström Macroglobulinemia
Waldenström macroglobulinemia (WM) is a lymphoplasmacytic lymphoma of B lymphocytes characterized by elevated levels of circulating IgM. Mutations in MYD88 and CXCR4 genes have been described. Disease manifestations relate to hyperviscosity and vascular complications resulting from the circulating paraprotein, as well as from neoplastic lymphoplasmacytic infiltration of important structures such as the bone marrow, lymph nodes, spleen, and other organs. Lymphadenopathy, hepatosplenomegaly, and anemia are characteristic.
Immunoglobulin M is responsible for some of the skin manifestations of the disorder. Elderly men are predominantly affected, and there is a strong familial predisposition. The cutaneous manifestations of WM can be divided into two categories: nonspecific and specific. Nonspecific manifestations are related to the hyperviscosity syndrome created by the circulating IgM and include purpura of the skin and mucous membranes. The purpura may be surmounted by tense giant bullae. Bleeding of the gums and epistaxis can occur. The IgM may behave as a cryoglobulin, resulting in purpura, livedo, cutaneous ulcerations, and vasculitis. Urticaria (some patients satisfy the diagnostic criteria for Schnitzler syndrome or progress from that disorder), disseminated xanthoma, and amyloid deposition can be seen. Specific skin lesions are of two types: specific skin deposits of aggregates of IgM (cutaneous macroglobulinosis) and cutaneous infiltrates with neoplastic lymphoid cells. The specific skin lesions usually occur once the diagnosis of WM is already known, but infrequently the skin lesions are the first clue to the diagnosis. The specific IgM deposits present clinically as subepidermal blisters (clinically and histologically resembling bullous amyloidosis) or translucent 1–3 mm papules. They are found most often on the lower extremity, even on the sole. Slight hyperkeratosis may be seen over the papules.
Histologically, the papules are composed of dermal nodular, homogeneous, and fissured pink deposits that tend to involve newly formed vessels. They are periodic acid–Schiff (PAS) positive but negative for Congo red. DIF identifies the dermal globules as being composed of IgM and is a useful diagnostic approach. When WM results in cutaneous lymphoid aggregates, the presentation is very nonspecific. Small, red-brown to violaceous macules, papules, nodules, or plaques may be present, usually on the face. Rosacea is often initially entertained as a diagnosis. Widespread skin involvement with a “deck-chair sign” (sparing the abdominal skinfolds) has been reported.
The natural history of WM is that of an indolent myelodysplasia. Treatment is directed at reducing the volume of neoplastic cells and should be managed by an oncologist. Most asymptomatic patients are followed or treated only when clinical disease occurs. Chlorambucil, cyclophosphamide, fludarabine, systemic corticosteroids, and rituximab or bortezomib, used alone or in combination, are initial therapeutic options. Rituximab, in combination with cyclophosphamide and dexamethasone, has been used as well. Rituximab is not used as a monotherapy in WM patients with hyperviscosity because they may experience a flare of their disease. Plasmapheresis can be effective in controlling acute symptoms of hyperviscosity syndrome. Ibrutinib has been shown to be safe and highly effective. With the emergence of monoclonal antibodies, proteasome inhibitors, and tyrosine kinase inhibitors, multiple targeted therapeutic options exist. Studies are ongoing for targeted agents to affect the MYD88 pathway.
Autier J, et al: Cutaneous Waldenström’s macroglobulinemia with “deck-chair” sign treated with cyclophosphamide. J Am Acad Dermatol 2005; 52: 45.
Camp BJ, Magro CM: Cutaneous macroglobulinosis. J Cutan Pathol 2012; 39: 962.
Castillo JJ, et al: Novel approaches to targeting MYD88 in Waldenstrom macroglobulinemia. Expert Rev Hematol 2017; 10: 739.
Chan I, et al: Cutaneous Waldenström’s macroglobulinaemia. Clin Exp Dermatol 2003; 28: 491.
Harnalikar M, et al: Keratotic vascular papules over the feet: a case of Waldenström’s macroglobulinaemia–associated cutaneous macroglobulinosis. Clin Exp Dermatol 2010; 35: 278.
Libow LF, et al: Cutaneous Waldenström’s macroglobulinemia. J Am Acad Dermatol 2001; 45: S202.
Oberschmid B, et al: M protein deposition in the skin: a rare manifestation of Waldenström macroglobulinemia. Int J Hematol 2011; 93: 403.
Paludo J, et al: Dexamethasone, rituximab, and cyclophosphamide for relapsed and/or refractory and treatment naïve patients with Waldenstrom macroglobulinemia. Br J Haematol 2017; 179: 98.
Tedeschi A, et al: Fludarabine plus cyclophosphamide and rituximab in Waldenström macroglobulinemia. Cancer 2012; 118: 434.
Yun S, et al: Waldenstrom macroglobulinemia. Clin Lymphoma Myeloma Leuk 2017; 17: 252.
Purpura Secondary to Clotting Disorders
Hereditary disorders of blood coagulation usually result from a deficiency or qualitative abnormality of a single coagulation factor, as in hemophilia or von Willebrand disease. Acquired disorders usually result from multiple coagulation factor deficiencies, as in liver disease, biliary tract obstruction, malabsorption, or drug ingestion. Acquired clotting disorders may also involve platelet abnormalities, as in DIC. Hemorrhagic manifestations are common and may be severe, especially in hereditary forms. Ecchymoses and subcutaneous hematomas are common, especially on the legs. Severe hemorrhage may follow trauma, and hemarthrosis is frequent. Other hemorrhagic manifestations include respiratory obstruction resulting from hemorrhage into the tongue, throat, or neck; epistaxis; gastrointestinal (GI) and genitourinary tract bleeding; and, rarely, CNS hemorrhage.
Carpenter SL, et al: Evaluating for suspected child abuse. Pediatrics 2013; 131: e1357.
Kruse-Jarres R, et al: Identification and basic management of bleeding disorders in adults. J Am Board Fam Med 2014; 27: 549.
Drug-Induced, Food-Induced, and Recreational Activity–Associated Purpura
Drug-induced purpura may be related to platelet destruction, vessel fragility, interference with platelet function, or vasculitis. Drug-induced thrombocytopenic purpura is discussed earlier in this chapter. Purpurogenic drugs include aspirin and other NSAIDs, allopurinol, thiazides, gold, sulfonamides, cephalosporins, hydralazine, phenytoin, quinidine, ticlopidine, and penicillin. Combinations of diphenhydramine and pyrithyldione can induce purpuric mottling and areas of necrosis. Cocaine-induced thrombosis with infarctive skin lesions is associated with skin popping. “Paintball” purpura has been described after recreational paintball contests. Hyperemesis or mechanical ventilation can cause a periorbital purpura, and anecdotal reports exist of patients performing “inversion” yoga developing periorbital purpura.
Topical EMLA cream can induce purpura within 30 minutes of application, a result of a toxic effect on the capillary endothelium. Purpura has been associated with the use of acetaminophen in patients with infectious mononucleosis. Small-vessel vasculitis, including urticarial vasculitis, has been caused by the ingestion of tartrazine dye. Pseudoephedrine can induce a pigmented purpura–like reaction. Patch testing reproduces the eruption. Purpuric contact dermatitis is rare and usually caused by rubber chemical or textile dyes.
Purpuric Agave Dermatitis
Agave americana is a large, thick, long-leaved, subtropical plant with a striking blue-gray color. It is often used in ornamental beddings in the southwestern United States. The plant grows up to 2 m ( ![]() feet) in diameter and may overgrow the surrounding landscape. These plants are deep rooted and difficult to remove, and some individuals attempt removal using a chainsaw. A striking purpuric dermatosis occurs in a pattern corresponding to the splatter of the plant’s sap. Histologically, there is vascular damage at the level of the capillary and postcapillary venule, with a sparse infiltrate of neutrophils and karyorrhectic debris, suggesting low-grade leukocytoclastic vasculitis. Papulovesicular lesions have also been described. The plant’s sap contains calcium oxalate crystals, as well as various acrid oils and saponins. The causative component is unknown.
feet) in diameter and may overgrow the surrounding landscape. These plants are deep rooted and difficult to remove, and some individuals attempt removal using a chainsaw. A striking purpuric dermatosis occurs in a pattern corresponding to the splatter of the plant’s sap. Histologically, there is vascular damage at the level of the capillary and postcapillary venule, with a sparse infiltrate of neutrophils and karyorrhectic debris, suggesting low-grade leukocytoclastic vasculitis. Papulovesicular lesions have also been described. The plant’s sap contains calcium oxalate crystals, as well as various acrid oils and saponins. The causative component is unknown.
Golfer’s and Exercise-Related “Vasculitis”
This syndrome occurs mostly in hot weather and affects primarily older men (>50). Golfing or exercise with prolonged walking is the trigger. The syndrome is characterized by asymptomatic or pruritic, burning, or stinging, purpuric, macular or slightly raised papules and plaques, predominantly just above the sock line near the ankles. Mild ankle swelling may be present. The lesions resolve in under 3 days in most patients. Histologically, true LCV (see discussion later in this chapter) is not seen, but erythrocytes and neutrophils are present in the affected tissue. About half the patients are taking antithrombotic agents. This syndrome probably represents a form of purpura caused by anticoagulation and prolonged erect posture rather than a true vasculitis.
Aboutalebi S, Stetson CL: Paintball purpura. J Am Acad Dermatol 2005; 53: 901.
Barabash-Neila R, et al: Agave americana causing irritant contact dermatitis with a purpuric component. Actas Dermosifiliogr 2011; 102: 74.
D’Addario SF, et al: Minocycline-induced immune thrombocytopenia presenting as Schamberg’s disease. J Drugs Dermatol 2003; 2: 320.
Diaz-Jara M, et al: Pigmented purpuric dermatosis due to pseudoephedrine. Contact Dermatitis 2002; 46: 300.
Genillier-Foin N, Avenel-Audran M: Purpuric contact dermatitis from Agave Americana. Ann Dermatol Venereol 2007; 134: 477.
Kelly RI, et al: Golfer’s vasculitis. Australas J Dermatol 2005; 46: 11.
Roldán-Marín R, de-la-Barreda Becerril F: Petechial and purpuric eruption induced by lidocaine/prilocaine cream. J Drugs Dermatol 2009; 8: 287.
Sagdeo A, et al: Purpuric eruption on the feet of a healthy young woman. JAMA Dermatol 2013; 149: 751.
Sanyal S, et al: Golfer’s purpura. J Eur Acad Dermatol Venereol 2016; 30: 1403.
Verma GK, et al: Purpuric contact dermatitis from footwear. Contact Dermatitis 2007; 56: 362.
Purpura Fulminans
Purpura fulminans is a rapidly progressive and fatal syndrome of intravascular thrombosis, circulatory collapse, and cutaneous infarction. Also known as purpura gangrenosa, there are three forms of the disease:
- 1.
Infectious (associated with bacterial or viral infection and DIC)
- 2.
Neonatal/hereditary (deficiency of protein C or S, or antithrombin III)
- 3.
Idiopathic (generally following a febrile illness, leading to acquired protein S deficiency)
The most common form is that associated with an infectious illness, usually bacterial septicemia (most often meningococcemia) but sometimes a viral infection (varicella). Asplenic patients, who are at risk for pneumococcal or meningococcal sepsis, are predisposed to purpura fulminans. Neonates with homozygous protein C or protein S deficiencies may have purpura fulminans ( Fig. 35.10 ). Some patients develop transient deficiencies of proteins C and S in response to infection. A number of reported cases of purpura fulminans have been associated with infections and factor V Leiden mutation, with normal protein C and protein S levels. Meningococcemia, streptococcal sepsis, Capnocytophaga sepsis (from a dog bite), staphylococcal septicemia, and urosepsis are the most common causes. Rickettsial disease and malaria may present as purpura fulminans. Active human herpesvirus 6 (HHV-6) replication with acquired protein S deficiency and purpura fulminans has been described.