Abstract
This chapter discusses the unique group of disorders known as the histiocytoses, with an emphasis on Langerhans cell histiocytosis (LCH). Both LCH and the group of disorders comprising non-Langerhans cell histiocytosis (non-LCH) range in severity from a mild, self-resolving skin-limited disease to a severe multisystem disease with a high risk of mortality. The chapter focuses primarily on the clinical features, pathogenesis, and treatment of these disorders allowing for easy comparison of these closely related conditions.
Keywords
BRAF V600E mutation, Juvenile xanthogranuloma, Langerhans cell histiocytosis (LCH), Langerin, Letterer–Siwe disease, Malignant histiocytosis, Non-Langerhans cell histiocytosis (non-LCH), Rosai–Dorfman disease
- •
Langerhans cell histiocytosis (LCH) is a clonal neoplastic disorder.
- •
LCH represents a spectrum of disease from asymptomatic skin-limited self-resolving lesions to systemic multisystem disease with high mortality.
- •
The majority of LCH cases harbor the BRAF V600E mutation, suggesting a potential role for BRAF inhibitors as treatment.
- •
LCH cells express S100, CD1a, CD207 (Langerin), and Fascin.
- •
The LCH-III trial confirmed a combination of vinblastine and prednisone as effective therapy for multisystem LCH both with and without risk of organ involvement.
- •
The cells in the non-LCH disorders express CD68 and CD163 and are always negative for Langerin (CD207).
- •
Juvenile xanthogranuloma is by far the most common of the non-LCH disorders.
- •
Leukemia, lymphoma, or a paraproteinemia are observed, sometimes frequently, in association with many of the non-LCH disorders.
Introduction
This chapter focuses on disorders commonly referred to as the “histiocytoses.” These conditions can be unpredictable and present in a wide variety of ways, from harmless self-resolving skin lesions to systemic disease with a high mortality rate. According to the Writing Group of the Histiocyte Society in 1987, the histiocytoses can be broadly classified into three groups: Langerhans cell histiocytosis (LCH, class I), non-Langerhans cell histiocytosis (non-LCH, class II), and malignant histiocytosis (class III).
Histiocytes of cutaneous importance are derived from bone marrow CD34+ progenitor cells. These cells can mature into Langerhans cells (which reside in the epidermis) or alternatively into monocytes, macrophages, or dendritic cells (which reside in the dermis or deeper soft tissues). Lesional tissue of LCH is composed of LCH cells (which have a similar immunophenotype to Langerhans cells) while the non-LCH disorders are comprised of monocytes, macrophages, and/or dendritic cells.
The malignant histocytic disorders are very rare and most are aggressive. These will be discussed briefly at the conclusion of this chapter.
Class I: Langerhans Cell Histiocytosis
Pathogenesis
The pathogenesis of LCH was uncertain until recently and there was much debate as to whether to classify LCH as a reactive or neoplastic condition. Studies have not shown evidence of an infectious etiology, and demonstrated elevated levels of multiple cytokines and interleukins in LCH are believed to be produced by the LCH inflammatory cell infiltrate rather than being responsible for causing the disease.
The recent major discovery of recurrent BRAF 600E mutations provides strong evidence to regard LCH as a neoplastic condition. Rare cases of familial LCH have been documented as well, and several older studies have demonstrated consistent clonality in LCH tissue. Given these later findings, LCH appears to represent a clonal neoplastic disorder.
Clinical Manifestations
Langerhans cell histiocytosis (LCH) is a rare disorder. Although it most commonly presents in children from ages 1 to 3 years it can occur at any age and is more common in males. The disease ranges from mild asymptomatic involvement of a single organ to severe, multiorgan system involvement with a high mortality. Given the wide range of disease expression, it is not surprising that the etiology of the disease was uncertain until later in the twentieth century, with four previously described syndromes found to represent overlapping clinical variants of LCH. These variants include Letterer–Siwe disease, Hand–Schuller–Christian disease, eosinophilic granuloma, and Hashimoto–Pritzker (congenital self-healing reticulohistiocytosis). While it is well recognized that LCH is one disease with a broad clinical spectrum, it is still helpful to note the older variants for perspective.
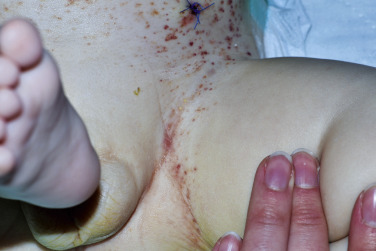
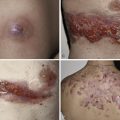
The acute form of LCH with diffuse and multisystem involvement was previously referred to as Letterer–Siwe disease. While it usually presents in infants less than 2 years of age, it can occur at any age. It presents as erythematous to skin-colored crusted 1 to 2-mm vesiculopustules, sometimes with secondary impetiginization, involving the scalp ( Fig. 22-1 ), neck, intertriginous areas, and bathing trunk area ( Fig. 22-2 ). Coalescing areas can become fissured, and petechia and purpura are also common. Soft tissue nodules can occur and rare presentations include ulcerative lesions and a papular form imitating molluscum contagiosum. Nail changes have also been described. The lungs, liver, spleen, and bone are commonly involved during the disease course. Lytic bone lesions are painful and primarily affect the cranium, vertebrae, and flat bones. So-called risk organ involvement includes the marrow (hematopoietic system), liver, lungs, and spleen. The prognosis of this form of LCH is variable. In those patients with risk organ involvement, especially in children less than 2 years of age, there is a high risk of mortality and systemic treatment is required.
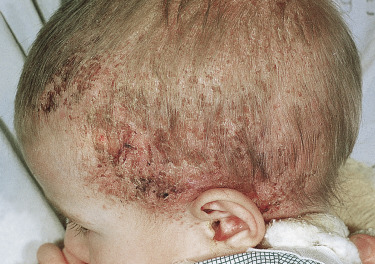
Some patients present with a chronic and progressive form of disease. This form most often presents during childhood. Although LCH in adults is very uncommon, a chronic progressive course is a common mode of presentation in adults. Bone lesions are very frequent in this form (at least 80%) and approximately one-third of patients develop skin or mucous membrane lesions. LCH cells also infiltrate the posterior pituitary gland in approximately 30% of patients causing diabetes insipidus. Rarely, exophthalmos develops as a late finding. The triad of diabetes insipidus, bone lesions, and exophthalmos was previously referred to as Hand–Schuler–Christian disease. The skin lesions, favoring the scalp, upper trunk, and intertriginous areas, are initially similar to those described in the acute form, but ulcerating nodules can develop especially in the gingival and genital areas. Lesions can occur in crops, older lesions can become xanthomatous, and some may spontaneously regress leaving scars. The cranium is the most common site of bone involvement. Chronic otitis media is also common and can be a clue to the presence of cranial involvement. Although patients with this variant tend to have persistent or progressive disease, most will survive with treatment.
Eosinophilic granuloma is a localized variant of LCH with a striking predilection for the bone. It most commonly occurs in older children. The most common presentation is a single, often asymptomatic granulomatous cranial lesion, though any bone can be affected. The lung can also be involved, which may be confused with but likely overlaps with a separate variant of LCH, pulmonary Langerhans cell histiocytosis (PLCH). Development of PLCH has a strong association with cigarette smoking and usually occurs in young adult males, with a predilection for Caucasian men. In eosinophilic granuloma, skin and mucous membrane lesions are rare although mucocutaneous nodules can present in the periorofacial, genital, or perianal regions. Patients with this variant can have a prolonged course but have an overall good prognosis.
Congenital self-healing reticulocytosis (Hashimoto–Pritzker) presents in newborn infants as a solitary lesion, multiple, or diffuse 2-mm to several centimeter red-brown papules or nodules, which may, over time, ulcerate and crust. Vesicular lesions can also occur. The vast majority of cases are limited to the skin but, uncommonly, involvement of the liver, colon, marrow, and spleen have been described. The prognosis is excellent as the lesions spontaneously regress over 2 to 3 months. However, postlesional scars can be permanent sequelae.
Differential Diagnosis
The differential diagnosis is extensive and includes seborrheic dermatitis, eczema, diaper dermatitis, Darier’s disease, urticaria pigmentosa, arthropod bites, scabies, leukemia, B- and T-cell lymphomas, and some of the non-Langerhans cell histiocytoses (especially indeterminate cell histiocytosis, benign cephalic histiocytosis, generalized eruptive histiocytosis, and xanthoma disseminatum). Ear involvement can easily be mistaken for chronic otitis media or otitis externa. As this is predominantly a disease of the pediatric population, it may not be suspected in adults leading to delay in diagnosis.
Histopathologic Findings
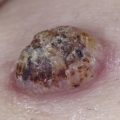
In the skin, an infiltrate of LCH cells is most often observed in the papillary dermis ( Fig. 22-3 ), but can also present in a nodular fashion in the dermis or in a perifollicular distribution. The epidermis is also typically infiltrated by LCH cells, sometimes giving a pagetoid appearance. LCH cells are large, and often have a reniform-shaped nucleus with abundant eosinophilic cytoplasm. The infiltrate commonly occurs along with eosinophils (sometimes numerous) and lymphocytes, and sometimes scattered plasma cells, mast cells, and neutrophils. Giant cells can occasionally be observed. LCH cells are confirmed by their expression of CD1a, S100, and Langerin (CD207), and their typical lack of expression for CD68, CD163, and factor XIIIa.
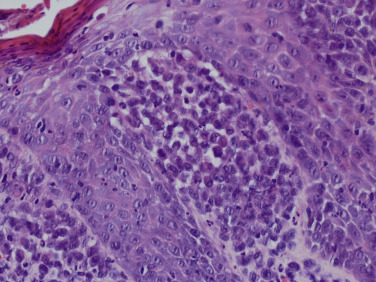
It is important for the dermatopathologist, dermatologist and nondermatologist clinician to recognize that reactive conditions can occasionally result in collections of normal Langerhans cells developing in the skin, usually in the context of mixed lymphohistiocytic inflammation with eosinophils. Examples include nodular scabies, other arthropod bite reactions, and atopic dermatitis.
Evaluation
In patients with biopsy-confirmed disease, complete blood count, liver function tests, routine metabolic panel, chest X-ray, and bone films as clinically indicated should be obtained. Additional evaluation of the CNS and bone marrow may also be necessary depending on the patient signs and symptoms and the severity of the disease.
Treatment
For patients with mild single-system involvement, conservative treatment is most often appropriate, and sometimes no treatment is needed if the patient is not symptomatic. For the skin, narrow-band ultraviolet therapy, topical corticosteroids, topical nitrogen mustard, or imiquimod cream are common options. For isolated symptomatic bone lesions, curettage, intralesional corticosteroid therapy, nonsteroidal anti-inflammatory drugs, or radiation are typical choices.
The Histiocyte Society Evaluation and Treatment Guidelines note that systemic therapy is suggested in several settings. It is indicated for patients with multisystem LCH, or single-system LCH with multifocal bone lesions, with “special site” lesions (vertebral with intraspinous extension or craniofacial bone with soft tissue involvement), or with “CNS risk” lesions. A combination of vinblastine and prednisone is the typical regimen for multisystem LCH. There are various treatment options for nonresponding patients with treatment tailored to the patient’s particular situation. The website for the Histiocyte Society ( www.histiocytesociety.org ) should be consulted for information on current trials and treatment guidelines.
Class II: Non-Langerhans Cell Histiocytosis
Pathogenesis and Pathology
The disorders that comprise non-Langerhans cell histiocytosis (non-LCH) include a large group of conditions, many of which involve the skin. There is clinical overlap between several of these disorders, notably between benign cephalic histiocytosis, juvenile xanthogranuloma, and generalized eruptive histiocytoma. The historical exclusion of some other histiocytic disorders (i.e., granuloma annulare and sarcoidosis) from the category appears somewhat arbitrary.
Most of these conditions are rare and the pathogenesis is unknown. A few partial exceptions exist. A familial form of Rosai–Dorfman disease, caused by mutations in the SLC29A3 gene, is now generally accepted. In necrobiotic xanthogranuloma, there is such a strong association with monoclonal gammopathies that a link seems likely. Finally, there appears to be a genetic link between leukemia (acute myelomonocytic leukemia and chronic myelomonocytic leukemia) and some cases of generalized eruptive histiocytoma.
Histologically, all of the non-LCH disorders are composed of histiocytes that stain positive for CD68 or CD163, and with just rare exceptions are negative for S100 and CD1a. Specifically, indeterminate cell histiocytosis stains positive with CD1a and S100, and Rosai–Dorfman stains positive with S100. The lack of staining with CD1a and with S100 in the non-LCH disorders are important discriminators from LCH, which clinically can mimic some of the non-LCH disorders. Although there is significant histologic overlap between the non-LCH disorders, there are subtle histologic findings often unique to the various non-LCH disorders that can assist in diagnosis. The detailed specific histologic features of these disorders are beyond the scope of this chapter.
Clinical Manifestations
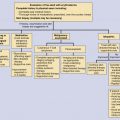
When learning and organizing these disorders, it is most useful to place them in categories. In this chapter ( Table 22-1 ), we have categorized them as follows: (1) primarily skin limited, (2) frequently mixed skin and systemic findings, and (3) systemic findings with infrequent or absent skin involvement. Table 22-1 provides the important features of these disorders allowing for quick comparisons.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


