Abstract
Cutaneous xanthomas may signal an underlying lipid, metabolic, or hematologic abnormality, so accurate diagnosis is essential. The clinical presentations of different types of xanthomas including eruptive, tuberous, planar, and tendinous xanthomas and their associated disease associations are discussed here along with a review of primary and secondary hyperlipoproteinemias.
Keywords
Dyslipoproteinemia, Eruptive, Planar, Tendinous, Tuberous, Xanthelasma, Xanthoma
- •
Cutaneous xanthomas present as yellow papules, nodules, or plaques and can signal the presence of an underlying lipid, metabolic, or hematologic abnormality.
- •
Xanthelasma is the most common cutaneous xanthoma and can be seen in association with dyslipoproteinemia in about half of patients.
- •
Xanthomas can also occur in normolipemic patients and herald an underlying metabolic, neurologic, or hematologic disorder.
- •
Treatments are available for many of the dyslipoproteinemia syndromes. Accurate dermatologic diagnosis and referral to a specialist is critical to mitigate against the predisposition for atherosclerotic, pancreatic, and other systemic comorbidities.
Cutaneous xanthomas result from intracellular and dermal deposition of lipids and can be a harbinger of underlying systemic disorders, most commonly hyperlipoproteinemias. Multiple forms of cutaneous xanthomas exist. While not entirely specific, different morphologies can point toward particular forms of primary hyperlipoproteinemia (see Table 26-1 ), or secondary hyperlipoproteinemias (see Table 26-2 ), or other normolipemic conditions. Many of the primary hyperlipoproteinemias are now defined at a molecular level with specific apoprotein or receptor mutations. A basic understanding of lipid metabolism provides insight into the pathogenesis of hyperlipoproteinemias.
| Elevated Lipoprotein Class | Fitzpatrick Type, Synonyms, and Primary Genetic Disorders | Lipid Profile | Cutaneous Xanthoma | Systemic Manifestations |
|---|---|---|---|---|
| Chylomicrons | Type I, familial lipoprotein lipase deficiency/Bürger–Grütz disease, familial apoprotein CII deficiencies | Hypertriglyceridemia | Eruptive | Presents in childhood or adolescence. Pancreatitis, lipemia retinalis. No increased risk of coronary artery disease |
| Chylomicrons and VLDLs | Type V, familial combined hyperlipidemia | Hypertriglyceridemia | Eruptive | Presents in adulthood. Association with diabetes, alcohol intake, obesity |
| VLDLs | Type IV, endogenous familial hypertriglyceridemia | Hypertriglyceridemia | Eruptive | Presents in adulthood. Association with diabetes, alcohol intake, obesity |
| LDLs | Type IIa, familial hypercholesterolemia, LDL receptor defect, defective B100/E, PCSK9 mutations | Hypercholesterolemia | Tendinous, tuberoeruptive, tuberous, planar (xanthelasma, intertringinous, interdigital web spaces ∗ ) | Atherosclerosis. Homozygous forms presents in childhood |
| LDLs and VLDLs | Type IIb, familial multiple lipoprotein-type hyperlipidemia, combined hyperlipidemia | Hypercholesterolemia | Tuberous, planar | Atherosclerosis, diabetes |
| IDLs | Type III, remnant hyperlipidemia, familial dysbetalipoproteinemia, broad beta deficiency, ApoE deficiency | Hypertriglyceridemia Hypercholesterolemia | Tuberoeruptive, tuberous, tendinous, planar, (xanthoma, striatum palmares—most characteristic) | Atherosclerosis |
| Diabetes mellitus |
| Cholestasis: Primary biliary cirrhosis, Alagille syndrome |
| Biliary atresia |
| Hypothyroidism |
| Nephrotic syndrome |
| Pregnancy |
| Alcoholism |
| Drugs: Estrogens, systemic retinoids, olanzapine, azacitidine, corticosteroids, antiretrovirals |
| Paraproteinemias: Multiple myeloma, lymphoma |
Lipid Metabolism and Primary Hyperlipoproteinemias
Lipids are a heterogeneous group of fats or fat-like substances that are insoluble in water, thus most plasma lipids are complexed as a lipoprotein with a hydrophilic phospholipid and apoprotein shell. Lipoproteins are classified by their density, which is a reflection of the core lipid content. Chylomicrons and very-low-density lipoproteins (VLDLs) have high triglyceride (TG) and low cholesterol ester (CE) content, while low-density lipoproteins (LDLs), intermediate-density lipoproteins (IDLs), and high-density lipoproteins (HDLs) have increasing CE and decreasing TG content. The lipoprotein structure allows the delivery of TG and CEs to peripheral cells for metabolic functions via interaction between apoproteins and specific receptors. An example is the interaction of the B100/E apoprotein found on VLDLs, IDLs, and LDLs and lipoprotein lipase on hepatocytes and capillary endothelium.
Two major pathways of lipoprotein synthesis exist: exogenous (dietary) and endogenous (hepatic production). In the endogenous pathway, ingested TGs are taken up in the intestine and packaged with a small amount of CEs into the central core of a chylomicron. These chylomicrons then enter the systemic circulation and release free fatty acids (via hydrolysis of the TGs) to the peripheral tissues. This process is mediated via the interaction between apoprotein CII on the chylomicrons and lipoprotein lipase on capillary endothelium. After release of the free fatty acids a chylomicron remnant is taken up by the liver via the apoprotein B100/E receptor.
In the endogenous pathway the liver forms VLDLs from circulating free fatty acids and hepatic TG stores. The rate-limiting enzyme is HMG-CoA, the target of the statin class of antihypercholesterolemia agents. VLDLs enter the circulation and after removal of TG content (via interaction of apoprotein CII on VLDL and lipoprotein lipase on the endothelium), an IDL is formed. IDLs are taken up by the liver or remain in circulation and become LDLs. LDLs deliver cholesterol to peripheral tissues for use in synthesis of cell membrane bilayers, myelin nerve sheaths, and for steroidogenesis and bile acid production. LDLs are ultimately taken up by the liver via apoprotein B100/E. HDLs transfer excess CEs from peripheral tissues and transfer them to other lipoproteins (LDLs, VLDLs, or chylomicrons) for transportation back to the liver. The clinical emphasis on low levels of LDL and high levels of HDL reflects appropriate levels of production and removal of cholesterols in the circulation.
Primary Hyperlipoproteinemias
Various classification schemas have been proposed for the primary hyperlipoproteinemias. The initial numerical schemes by Frederickson and Lee in 1965 are based on the serum lipoproteins present. Numerous synonyms exist and today this classification scheme is enhanced by insights from molecular biology (see Table 26-1 ).
Hyperchylomicronemia
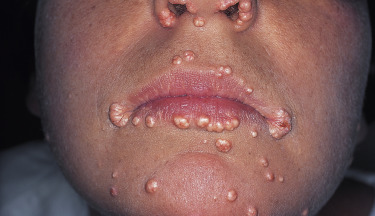
Two genetically determined defects of TG removal lead to hypertriglyceridemia and hyperchylomicronemia: these are autosomal recessive lipoprotein lipase deficiency (also known as type I or Bürger–Grütz disease) and familial apoprotein CII deficiency. However, the majority of patients with high levels of chylomicrons and TGs have acquired secondary forms of hyperlipidemia. Pancreatitis and bouts of abdominal pain are common in severe type I disease, often beginning in early childhood. In addition, these children develop hepatosplenomegaly, eruptive xanthomas, and lipemia retinalis, especially when the TG levels exceed 4000 mg/dL. Premature atherosclerotic vascular disease does not occur in type I disease. Patients with an absence of lipoprotein lipase activator (apoprotein CII) first develop symptoms after adolescence. Patients with type V disease have elevations of both chylomicrons and VLDLs—so-called familial combined hyperlipidemia. The symptoms usually begin in adult life, and as is true for many patients with primary hyperlipidemia, secondary factors, such as alcohol intake, obesity, associated renal disease, or diabetes mellitus, are frequently involved in exacerbation of the disease.
Increased VLDLs
Endogenous familial hypertriglyceridemia (type IV disease) results primarily from accelerated production of VLDL in the liver. This autosomal dominant disorder is common. The symptoms first appear in adulthood, frequently being precipitated by the ingestion of large amounts of carbohydrate or alcohol. Patients with this disorder are often obese, diabetic, and hyperuricemic and have an increased risk of coronary artery disease. Eruptive xanthomas are common, and xanthoma striatum palmare can also occur. VLDLs may be elevated along with chylomicrons in type V disease. Patients with elevations of both VLDL and LDL have type IIb disease.
Increased LDLs
LDLs alone are elevated in type IIa disease (familial hypercholesterolemia), and are elevated together with VLDLs in type IIb disease. There are several different phenotypic genetic conditions of familial hypercholesterolemia, and the severity of the clinical manifestations varies considerably. Xanthomas, especially the tendinous and tuberous types, are prominent, and there is a significant increase in the incidence of coronary artery disease, often beginning in early adulthood.
Elevated IDLs
Patients with high levels of cholesterol and TGs, carried in remnant lipoproteins (IDLs), have type III or broad-β disease (familial dysbetalipoproteinemia). Type III hyperlipoproteinemia is inherited as an autosomal dominant disease, although similar remnant lipoprotein accumulation in the plasma has been seen as a secondary phenomenon in hypothyroidism and multiple myeloma. The disorder appears to be related to a defect in the removal of these remnants from the circulation. Clinically, patients with broad-β disease are usually obese, glucose- intolerant, and have cutaneous xanthomas and atherosclerotic disease.
Secondary Hyperlipoproteinemias
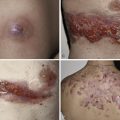
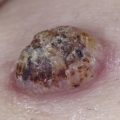
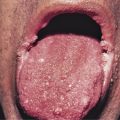
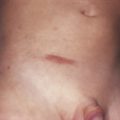
The majority of cases of xanthomatosis are secondary, rather than the primary familial disorders listed in Table 26-1 . Secondary hyperlipidemias result from disease in various organs (e.g., liver, kidney, thyroid, or pancreas) and are caused by a disturbance in the metabolism of TGs and cholesterol ( Table 26-2 ). Eruptive xanthomas may appear when hypertriglyceridemia develops in patients with uncontrolled diabetes mellitus, and in patients with the nephrotic syndrome. Tuberous and eruptive xanthomas can be seen in patients with hypothyroidism, but only rarely. Infants with biliary atresia and adults with biliary cirrhosis may develop any of the four types of xanthoma ( Figs. 26-1 and 26-2 ). Diffuse plane xanthomas are associated primarily with malignancies of the reticuloendothelial system, including multiple myeloma and lymphoma. Associated dyslipoproteinemias have been reported and attributed to complexes between the lipoproteins and paraproteins.