Abstract
Malignant lymphomas may affect the skin both primarily (i.e., without extracutaneous disease at presentation) and secondarily (i.e., as specific manifestations of a primary extracutaneous—usually nodal—lymphoma). Differentiation of primary from secondary cutaneous lymphomas is paramount, as staging investigations, treatment modalities, and prognosis are different. In some cases, only accurate staging investigations allows differentiation of primary cutaneous lymphomas from their nodal counterparts, as clinical, histological, and phenotypic features may be similar. Primary cutaneous lymphomas represent a heterogeneous group of entities with either a T-cell or a B-cell phenotype. The most frequent primary cutaneous lymphoma is by far mycosis fungoides, representing approximately 50% of all cutaneous lymphomas. The World Health Organization classification of hematological malignancies lists most primary cutaneous lymphomas as separate entities. In extranodal lymphomas, the skin may be involved secondarily by neoplastic lymphocytes. In general, specific skin manifestations of extracutaneous lymphomas retain the histopathologic and phenotypic features of the original tumor. Dedifferentiation or blastic progression, however, may be observed in some cases (e.g., in cutaneous Richter’s syndrome, representing in many cases large cell transformation of B-cell chronic lymphocytic leukemia—B-CLL). In these instances, the cutaneous manifestations show different morphology and phenotype compared to the original extracutaneous tumor. Besides specific skin manifestations the skin may be the site of several non-specific symptoms and/or disorders (e.g., generalized pruritus, Sweet’s syndrome, pyoderma gangrenosum, etc.).
Keywords
Blastic plasmacytoid dendritic cell neoplasm, Cutaneous B-cell lymphomas, Cutaneous diffuse large B-cell lymphoma, Cutaneous follicular lymphoma, Cutaneous marginal zone lymphoma, Cutaneous T-cell lymphomas, Leg-type, Mycosis fungoides, Primary cutaneous CD30+ lymphoproliferative disorders, Sézary syndrome
- •
Cutaneous lymphomas represent a heterogeneous group of malignant lymphomas arising primarily in the skin.
- •
Most cutaneous lymphomas are listed as separate entities in the World Health Organization classification of hematologic malignancies.
- •
Differentiation of primary cutaneous lymphomas from extracutaneous lymphomas with secondary skin manifestations is paramount, as management of patients is different.
- •
In some cases, only accurate staging investigations allow differentiation of primary cutaneous lymphomas from their nodal counterparts, as clinical, histological, and phenotypic features may be similar.
- •
In extranodal lymphomas, besides specific skin manifestations the skin may be the site of several nonspecific symptoms and/or disorders (e.g., generalized pruritus, Sweet’s syndrome, pyoderma gangrenosum, etc.).
Malignant lymphomas may affect the skin both primarily (i.e., without extracutaneous disease at presentation) and secondarily (i.e., as specific manifestations of a primary extracutaneous—usually nodal—lymphoma). Differentiation of primary from secondary cutaneous lymphomas is paramount, as staging investigations, treatment modalities, and prognosis are different. In fact, the World Health Organization (WHO) classification of hematologic neoplasia recognizes several types of primary cutaneous lymphomas, the most frequent being mycosis fungoides (MF) ( Table 20-1 ). In addition to specific skin manifestations of hematologic malignancies, many non-neoplastic cutaneous conditions may be variably associated with systemic lymphomas, the most important of them being briefly discussed at the end of this chapter and summarized in Table 20-2 .
| Mycosis Fungoides |
|---|
| Sézary syndrome Adult T-cell leukemia/lymphoma ∗ Primary cutaneous CD30+ lymphoproliferative disorders
Extranodal NK/T-cell lymphoma, nasal type ∗ Primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma Primary cutaneous gamma/delta T-cell lymphoma Primary cutaneous CD4+ small/medium T-cell lymphoma Extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue (MALT lymphoma) † Primary cutaneous follicle center lymphoma Primary cutaneous diffuse large B-cell lymphoma, leg type Intravascular large B-cell lymphoma ∗ Blastic plasmacytoid dendritic cell neoplasm ∗ |
∗ These entities are not always primary cutaneous, but may arise in the skin in the absence of systemic manifestations (i.e., negative staging at presentation).
† This category includes cases commonly classified as cutaneous marginal zone lymphoma, which in the WHO classification has been lumped together with other extranodal variants of the disease.
| Associated with Tumor-Induced or Treatment-Related Bone Marrow Suppression and Resulting Cytopenia |
| Pallor |
| Purpura, especially petechial |
| Gingival hemorrhage; epistaxis; prolonged bleeding after minor injuries |
| Oral ulcerations in neutropenic patients |
| Associated with either Tumor-Specific or Treatment-Related Immune Dysregulation |
| Opportunistic infections; unusual presentations of common infections (e.g., vegetating lesions, prolongued duration of self-limited disease, severe manifestations of otherwise banal infections) |
| Associated with Autoantibodies Produced by the Hematologic Neoplasm |
| Paraneoplastic pemphigus † |
| Associated with Deposition of Amyloid Protein |
| Cutaneous amyloidosis |
| Crystal storing histiocytosis |
| Associated with Paraproteinemia but without Deposition of M-protein |
| Scleromyxedema |
| Scleredema |
| Normolipemic plane xanthoma |
| Necrobiotic xanthogranuloma |
| POEMS syndrome |
| AESOP syndrome |
| Schnitzler’s syndrome |
| Other Conditions (Exact Mechanism and Pathogenesis of Skin Lesions Not Known) |
| Generalized idiopathic pruritus ‡ |
| Sweet’s syndrome (acute febrile neutrophilic dermatosis) § |
| Bullous pyoderma gangrenosum |
| Acquired ichthyosis |
| Cutaneous and/or systemic sarcoidosis || |
∗ Many of these conditions may be observed in different settings other than hematologic neoplasms; some are mostly, but not invariably, associated with an underlying lymphoproliferative disorder.
† Associated mostly with B-cell lymphomas and leukemias.
‡ May be observed in Hodgkin’s lymphoma (HL), as well as less frequently in other types of non-Hodgkin’s lymphoma (NHL); it may represent the first clinical manifestation of the disease.
§ As paraneoplastic disorder observed mostly in patients with myelogeneous leukemia and related conditions; skin lesions may harbor neoplastic cells of the underlying condition.
|| Sarcoidosis may antedate the diagnosis of NHL (“sarcoidosis-lymphoma syndrome”) or arise during the course of the disease.
Primary Cutaneous Lymphomas
Primary cutaneous lymphomas represent a heterogeneous group of lymphoproliferative disorders affecting the skin. By definition a primary cutaneous lymphoma does not show extracutaneous involvement at presentation (Sézary syndrome [SS] represents an exception, as involvement of the blood is a prerequisite for the diagnosis). Table 20-1 summarizes the entities included in the WHO classification. Precise diagnosis and distinction from similar entities involving the skin secondarily is crucial in order to manage patients properly.
Primary Cutaneous T-Cell Lymphomas
Mycosis Fungoides
MF represents by far the most common type of primary cutaneous T-cell lymphoma (CTCL) and of cutaneous lymphoma in general, accounting for almost half of all cases. It is defined as a tumor composed of small/medium-sized, epidermotropic T-helper lymphocytes. The disease is characterized by a chronic course with prolonged survival. The incidence is around 6–7 cases/10 6 , with regional variations and with a regular increase in recent decades. Although adults, especially elderly, are usually affected, MF represents the most common cutaneous lymphoma in children and adolescents as well.
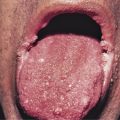
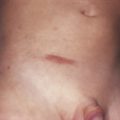
Clinically, lesions of MF can be divided morphologically into patches, plaques, and tumors. Morphology is commonly used to classify the disease in three clinical stages according to the presence of corresponding lesions (patch, plaque, and tumor stage), with a good correlation with prognosis (this is a good “rule of thumb” to discuss with patients and guides discussion of available treatment options, but of course a more precise staging system should be applied—see below). So-called plaque and tumor stages probably should be considered together as a more aggressive phase of the disease, compared to the chronic course of patch-stage disease. Pruritus is often a prominent symptom in all stages of the disease, and may be very difficult to treat. The diagnosis of MF is based mainly on clinicopathologic correlation. Immunohistochemical and molecular analyses offer adjunctive information that should be integrated with the clinicopathologic assessment.
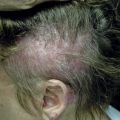
The precise characterization of the early phases of MF is still a matter of debate, and terms such as “parapsoriasis en plaques” or large plaque parapsoriasis are used differently to denote either a non-neoplastic condition that may progress to MF, or an early manifestation of the disease. In this stage the disease presents with variably large erythematous patches commonly located in sun-protected areas. Loss of elastic fibers and atrophy of the epidermis may confer on the lesions a wrinkled appearance. Notwithstanding the discussion on the nosology of “parapsoriasis en plaques,” patients with early MF should not undergo extensive staging investigations (only clinical examination with assessment of percentage of body involvement and of superficial lymph nodes), and should be managed in a nonaggressive way.
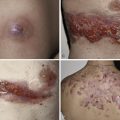
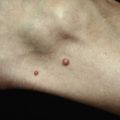
Plaques of MF are characterized by infiltrated, variably scaling, reddish-brown, indurated lesions. Typical patches are usually observed contiguous to plaques or at other sites of the body. Tumors in MF may be observed in the absence of other lesions, or in combination with patches and plaques. They may be solitary or, more often, localized or generalized. Tumors, as the sole manifestation of MF, except as relapsed disease are highly unusual, and a tumor d’emblee diagnosis would require exclusion of cutaneous anaplastic large cell lymphoma, or a nodule of lymphomatoid papulosis, or pleiomorphic small/medium CD4+ lymphoma, NK/T cell lymphoma, etc. Ulceration is common. Erythroderma can develop in the course of the disease, rendering distinction from SS difficult. Patients with infiltrated plaques, tumors, or erythroderma should be screened for extracutaneous involvement (laboratory investigations, sonography of lymph nodes, computed tomography and/or positron emission tomography scan of chest, abdomen and pelvis, bone marrow biopsy, examination of the peripheral blood).
Staging for MF is performed according to a system proposed by a joint working group of the International Society of Cutaneous Lymphoma (ISCL) and the European Organization for Research and Treatment of Cancer (EORTC) Cutaneous Lymphoma Task Force ( Table 20-3 ).
| Skin |
|
| Lymph Nodes |
|
| Visceral |
|
| Blood |
|
| Stage |
| IA T1N0M0B0,1 IB T2N0M0B0,1 |
| II T1,2N1,2M0B0,1 IIB T3N0–2M0B0,1 |
| III T4N0–2M0B0,1 IIIA T4N0–2M0B0 IIIB T4N0–2M0B1 |
| IVA1 T1–4N0–2M0B2 IVA2 T1–4N3M0B0–2 IVB T1–4N0–3M1B0–2 |
∗ For skin, patch indicates any size skin lesion without significant elevation or induration. Presence/absence of hypo- or hyperpigmentation, scale, crusting, and/or poikiloderma should be noted.
† For skin, plaque indicates any size skin lesion that is elevated or indurated. Presence or absence of scale, crusting, and/or poikiloderma should be noted. Histologic features such as folliculotropism or large cell transformation (>25% large cells), CD30+ or CD30−, and clinical features such as ulceration are important to document.
‡ For skin, tumor indicates at least one 1-cm diameter solid or nodular lesion with evidence of depth and/or vertical growth. Note total number of lesions, total volume of lesions, largest size lesion, and region of body involved. Also note if histologic evidence of large cell transformation has occurred. Phenotyping for CD30 is encouraged.
§ For node, abnormal peripheral lymph node(s) indicates any palpable peripheral node that on physical examination is firm, irregular, clustered, fixed, or 1.5 cm or larger in diameter. Node groups examined on physical examination include cervical, supraclavicular, epitrochlear, axillary, and inguinal. Central nodes, which are not generally amenable to pathologic assessment, are not currently considered in the nodal classification unless used to establish N3 histopathologically.
|| For viscera, spleen and liver may be diagnosed by imaging criteria.
¶ For blood, Sézary cells are defined as lymphocytes with hyperconvoluted cerebriform nuclei. If Sézary cells cannot be used to determine tumor burden for B2, then one of the following modified International Society of Cutaneous Lymphoma criteria along with a positive clonal rearrangement of the T-cell receptor may be used instead: (1) expanded CD4+ or CD3+ cells with CD4:CD8 ratio of 10 or more, (2) expanded CD4+ cells with abnormal immunophenotype including loss of CD7 or CD26.
# A T-cell clone is defined by polymerase chain reaction or Southern blot analysis of the T-cell receptor gene.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


