Abstract
Gastrointestinal diseases have many cutaneous associations. Gastrointestinal hemorrhage may be associated with the peau d’orange plaques of pseudoxanthoma elasticum or the macular telangiectases of hereditary hemorrhagic telangiectasia. Adenomatous polyposis may be associated with epidermal cysts, subcutaneous fibromas or dermoid tumors. Hamartomatous polyposis, including Peutz–Jeghers syndrome and Cowden’s syndrome may be associated with mucocutaneous pigmentation and trichilemmomas, respectively. Malabsorption syndromes include celiac disease with its classical findings of dermatitis herpetiformis, and acrodermatitis enteropathica with its classical findings of periorificial and acral dermatitis. Malabsorption of any type may be associated with skin signs of a wide variety of vitamin deficiency states. Inflammatory bowel disease findings include aphthosis, pyoderma gangrenosum, and erythema nodosum. Pancreatitis may present with panniculitis or purpura. Glucagonoma is classically associated with migratory necrolytic erythema. These associations as well as their pathogenesis and treatment are described in this chapter.
Keywords
Acrodermatitis enteropathica, Adenomatous polyposis, Aphthosis, Celiac disease, Cowden’s syndrome, Cullen’s sign, Dermatitis herpetiformis, Dermoid tumors, Epidermal cysts, Erythema nodosum, Fibromas, Gastrointestinal hemorrhage, Glucagonoma, Hamartomatous polyposis, Hereditary hemorrhagic telangiectasia, Inflammatory bowel disease, Malabsorption, Mat-like telangiectasia, Migratory necrolytic erythema, Mucocutaneous pigmentation, Pancreatitis, Panniculitis, Peau d’orange plaques, Peutz–Jeghers syndrome, Polyposis, Pseudoxanthoma elasticum, Pyoderma gangrenosum, Trichilemmomas, Turner’s sign, Vitamin deficiency, Zinc deficiency
- •
Gastrointestinal hemorrhage may be associated with pseudoxanthoma elasticum or hereditary hemorrhagic telangiectasia.
- •
Adenomatous polyposis may be associated with epidermoid tumors, fibromas, and dermoid tumors.
- •
Peutz–Jeghers syndrome and Cowden’s syndrome are associated with hamartomatous polyps.
- •
Malabsorption may be associated with dermatitis herpetiformis and celiac disease or acrodermatitis enteropathica and zinc deficiency.
- •
Inflammatory bowel disease is associated with neutrophilic dermatoses such as pyoderma gangrenosum, aphthosis, and Sweet’s syndrome as well as erythema nodosum.
- •
Pancreatitis may be manifested as purpura in specific areas or as panniculitis.
There are a number of diseases of the gastrointestinal tract that feature recognizable cutaneous diseases as part of their spectrum. This discussion includes cutaneous associations of the following disorders: gastrointestinal hemorrhage, polyposis, malabsorption, inflammatory bowel disease, and pancreatic disease. The genetic basis of many of these diseases has now been elucidated ( Table 29-1 ).
| Pseudoxanthoma elasticum: ATP-binding cassette transporter C6 ( ABCC6 ) |
| Hereditary hemorrhagic telangiectasia 1: endoglin ( ENG ) |
| Hereditary hemorrhagic telangiectasia 2: activin A receptor, type II-like kinase 1 ( ACVRL1 ) |
| Gardner’s syndrome: adenomatous polyposis coli ( APC ) |
| Peutz–Jeghers syndrome: serine/threonine kinase ( STK11 ) |
| Cowden’s disease: phosphatase and tensin homolog ( PTEN ) |
| Acrodermatitis enteropathica: solute carrier family 39 (zinc transporter) member 4 ( SLC39A4 ) |
Gastrointestinal Hemorrhage
Extensive gastrointestinal hemorrhage may occasionally be related to systemic disorders that are easily recognized by their cutaneous findings. Pseudoxanthoma elasticum and hereditary hemorrhagic telangiectasia will be discussed.
Pseudoxanthoma Elasticum
Pseudoxanthoma elasticum (PXE) is a rare inherited disorder of elastic tissue that occurs in 1:25,000–100,000 births. There is progressive calcification of tissue rich in elastin fibers, including the skin, retina, and blood vessels. There is significant heterogeneity in the age of onset as well as the severity of organ system involvement. The greatest morbidity lies in reduced vision from macular hemorrhage and scarring of the macula. Affected individuals have a normal lifespan.
Pathogenesis
PXE is inherited in an autosomal recessive manner. PXE is caused by mutation in the ABCC6 gene, ATP-binding cassette transporter protein. The gene has been mapped to chromosome 16p13.1. At least one ABCC6 mutation can be found in 80% of affected individuals. ABCC6 protein is largely expressed in the liver and kidney. The ABCC6 gene encodes for a cellular transport protein, giving rise to the concept that PXE may be a systemic metabolic disorder rather than purely a structural disorder of connective tissue. The elastic fibers are abnormal in the affected tissues. Changes include fragmentation and calcification of degenerated elastic tissue fibers in the middle and deep reticular dermis. Progressive accumulation of calcium within the elastic fibers leads to fracture and destruction.
Presentation
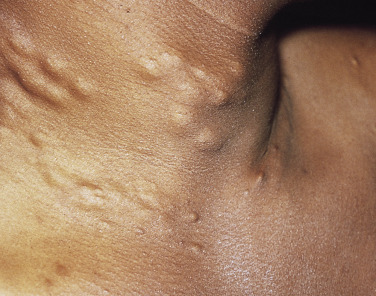
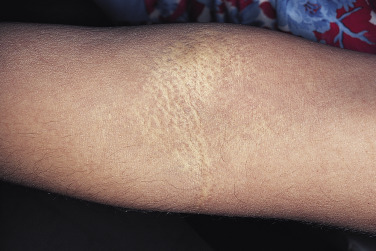
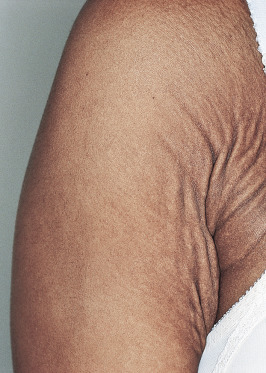
Cutaneous findings usually begin in the second to third decades. Calcification of elastic fibers results in yellowish discoloration of the skin. Affected skin reveals progressive yellowish coalescent papules on the lateral aspect of the neck, the flexural creases of the antecubital fossa and popliteal fossa, the axilla, and the groin. These yellow papules have been described as having a “plucked chicken skin” appearance. In severe cases, the skin appears loose and wrinkled ( Figs 29-1 and 29-2 ).
The earliest ocular finding is diffuse mottling of the fundus. In the second to third decade, angioid streaks develop in the eye. These present as linear and branching networks of grayish discoloration radiating from the optic disc. Angioid streaks are larger in caliber than blood vessels and represent choroidal neovascularization in the elastic lamina of Bruch’s membrane of the retina. Angioid streaks do not affect visual acuity. Subretinal neovascularization and hemorrhage lead to scarring and loss of vision. If the macula is involved, the loss of vision becomes permanent.
Calcification of the elastic media of blood vessels results in hypertension, peripheral vascular disease, coronary artery disease, aneurysms, and cerebral hemorrhage. PXE affects the elastic tissue of the cardiac valves, the myocardium, and the pericardium. Gastrointestinal hemorrhage occurs in approximately 10% of patients with PXE. The most common site of bleeding is the stomach. This may develop from gastritis or peptic ulcer disease. Diffuse superficial erosions rather than focal bleeding are often found in the gastrointestinal tract. The bleeding is difficult to control due to defective vasoconstriction of the arteries. Gastric bleeding may occur early before ocular and cutaneous changes are fully developed.
Evaluation
The diagnosis is confirmed by the clinical picture and the demonstration of fragmented, calcified elastic fibers on skin biopsy that is essential for diagnosis. Skin biopsy of flexural skin or scars is warranted in both suspected cases and potentially involved family members. Examination by light microscopy demonstrates fragmentation and irregular clumping of elastic tissue in the middle to deep dermis. Staining for calcium frequently shows significant elastic tissue calcification. Although such findings are classically present in involved skin, clinically normal-appearing skin of the flexural areas may show similar findings. Biopsy may confirm a diagnosis of PXE in patients with angioid streaks and minimal cutaneous findings.
The diagnosis is suggested by angioid streaks in the second decade of life, or by a positive family history. Angioid streaks are not sufficient for the diagnosis. They are highly suggestive of the disease in patients with a positive family history. In 85% of patients with skin findings, angioid streaks are present in the eye grounds. If PXE is suspected, detailed examination by an ophthalmologist is essential. Because eye changes are seen early in life, funduscopic examination is also recommended for screening of relatives of known patients.
Recently, molecular genetic testing for the ABCC6 gene has become available. Testing detects a mutation in one allele in almost all affected individuals and in both alleles in close to 90%. Sequence analysis detects missense mutations, nonsense mutations, frameshift mutations, as well as small deletions and insertions. Genetic testing and interpretation of results should be accompanied by genetic counseling.
Differential Diagnosis
The characteristic yellowish papules of PXE may be confused with solar elastosis. The neck is a common site for both, but PXE also occurs in the axilla, groin, and popliteal and antecubital fossae. Solar elastosis produces abnormal elastic tissue that is described histologically as dense masses in the upper dermis. The abnormal elastic tissue of solar elastosis does not stain for calcium. PXE shows fragmented clumps of elastic tissue in the mid to lower reticular dermis. Besides solar elastosis, skin lesions similar to PXE are found in conditions such as Buschke–Ollendorf syndrome, late-onset focal dermal elastosis, and cutis laxa.
Angioid streaks are valuable markers for the diagnosis of PXE, but are not pathognomonic findings. Angioid streaks may be seen in numerous disorders ( Table 29-2 ), but are most commonly related to sickle cell anemia or Paget’s disease of the bone. No fundamental pathogenic relationship between the disorders causing angioid streaks has been established.
| Paget’s disease of the bone |
| Sickle cell anemia |
| Thalassemia |
| Ehlers–Danlos syndrome |
| Tuberous sclerosis syndrome |
| Sturge–Weber syndrome |
| Neurofibromatosis |
| Hemolytic anemia |
| Diabetes |
| Hemochromatosis |
| Hyperphosphatasemia |
| Hypercalcinosis |
| Lead poisoning |
| Pituitary disorders |
| Acromegaly |
| Myopia |
| Traumatic choroidal rupture |
Treatment
There is no specific treatment for the basic defect in PXE. Weight control, avoidance of smoking, and aggressive management of hypertension and lipid disorders may reduce vascular complications. Both aspirin and nonsteroidal anti-inflammatory drugs should be avoided. Affected individuals should be discouraged from contact sports. Hemorrhage and vascular occlusive disease with PXE are managed medically.
Successful surgical removal of redundant skin for cosmetic reasons has been reported. Complications included slow healing, extrusion of calcium particles through scars, and widening of surgical scars. The majority of patients were highly satisfied with the results.
Hereditary Hemorrhagic Telangiectasia
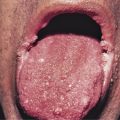
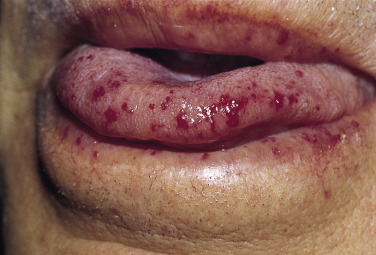
Hereditary hemorrhagic telangiectasia (HHT) is an autosomal dominant disorder characterized by vascular dysplasia. Telangiectases are permanent dilatations of capillaries that usually blanch when pressure is applied. These telangiectases begin on the mucous membranes of the nose and mouth during childhood. Telangiectases of HHT are best demonstrated by stretching the mucosal surface of the lower lip between the thumb and the forefinger, revealing 2- to 3-mm punctate red macules that may become papular with age ( Fig. 29-3 ). In young adults with HHT, an excess mortality has been attributed to HHT.
Pathogenesis
The prevalence of HHT is 1 per 10,000 of the population, which is much more common than previously thought. The mode of inheritance is autosomal dominant with a penetrance of approximately 97%. Two molecular subtypes of HHT are now recognized. Both HHT-1 and HHT-2 are multisystem vascular dysplasias caused by specific gene mutations found through linkage studies. The two subtypes reported have distinctions in the severity of disease and genetic markers.
HHT-1 is associated with a higher prevalence and increased severity of arteriovenous malformations. These patients have a higher risk of developing complex vascular abnormalities in the lungs and central nervous system at an early age. A mutation in the endoglin ( ENG ) gene has been described in HHT-1. Endoglin, which is a transforming growth factor-β (TGF-β)-binding protein, is expressed on capillaries, veins, and arteries. The ENG gene maps to chromosome 9q34.1.
HHT-2 is associated with a milder phenotype, reduced penetrance, and a later age of onset. Pulmonary arteriovenous malformations are less common in HHT-2 than in HHT-1. Activin A receptor, type II-like kinase 1 ( ACVRL1 ) is the mutated gene described in HHT-2. The ACVRL1 gene maps to chromosome 12q11-q14. ACVRL1 is detected in highly vascularized tissues and expressed primarily on endothelial cells.
Histopathologically, irregularly dilated capillaries and venules develop in the papillary dermis. There is a lack of perivascular support, including reduced pericytes, smooth muscle, and elastic fibers. Abnormally large collagen bundles with irregular banding have also been described. Vessels with defective perivascular support are especially sensitive to insult in the gastrointestinal tract, where the epithelium is not cornified.
Presentation
Telangiectases develop on the undersurface of the tongue and floor of the mouth at puberty. Spontaneous and recurrent epistaxis is the most common presentation in childhood. Vascular abnormalities of the gastrointestinal, pulmonary, and nervous systems develop after the fourth decade of life. Telangiectatic lesions in the gastrointestinal tract cause bleeding in 20% to 30% of patients. Onset of gastrointestinal bleeding is usually in the fourth to sixth decades. Peptic ulcer disease is common in HHT patients. Bleeding is frequently from telangiectatic mucosa that has spontaneously eroded. Gastrointestinal hemorrhage tends to be progressive. With advancing age, gastrointestinal bleeding leads to severe anemia. Potential fatal problems with bleeding warrant close attention.
Elevated transaminases, γ-glutamyl transferase, and alkaline phosphatase have been reported in up to 30% of patients with HHT. Arteriovenous malformations of the liver are likely if there is hepatomegaly or a bruit over the liver. Cirrhosis of HHT is described as abnormal dilated vessels and changing stroma throughout the liver. Bleeding from hepatic arteriovenous fistulae is rare.
Pulmonary arteriovenous fistulae have been found in 15% to 33% of patients. Up to 50% of all pulmonary arteriovenous fistulae are associated with HHT. Cyanosis, clubbing, and dyspnea are late signs of arteriovenous fistulae. Most lesions are detected with a combination of chest radiography and measurement of PaO 2 .
High cardiac output states secondary to severe anemia and systemic arteriovenous shunting may produce biventricular failure. Multiple arteriovenous fistulas may present with central nervous system findings, including transient ischemic attacks and cerebrovascular accidents. Cerebrovascular anomalies include arteriovenous malformation, capillary angiomas, and telangiectases. It is estimated that cerebral arteriovenous malformations occur in 5% to 10% of patients with HHT. Focal neurologic defects may result from these vascular malformations of the brain, spinal cord, and meninges. Patients with pulmonary and/or cerebral arteriovenous malformations risk early death from rupture of the diseased vessels.
Evaluation
The clinical diagnosis of HHT is based on the presence of telangiectases and on a family history of HHT. Four criteria for the diagnosis of HHT include epistaxis, telangiectases (lips, oral cavity, fingers, nose), visceral lesions, and a family history. The diagnosis is definitive with three or four criteria, but cannot be established with fewer than two. HHT lesions occur as multiple, 2- to 4-mm, usually symmetric, punctate, blanching macules, and as minimally elevated papules on the lips, face, nasal and oral mucosa, hands, feet, and upper extremities. The mode of inheritance is autosomal dominant, so a family history of bleeding is common.
If the characteristic telangiectases are present on the skin or mucous membranes, a detailed family history and history of bleeding episodes are essential. Clinical telangiectases may be few, especially in children and adolescents; therefore, close physical examination is necessary. Recurrent epistaxis at a young age may precede obvious telangiectases by many years, and this makes family history especially important. Examination of family members should focus on the wide spectrum of HHT.
Differential Diagnosis
There are a large number of diseases that can produce cutaneous vascular abnormalities. Telangiectatic mats with similar distributions appear in CREST syndrome (calcinosis, Raynaud’s phenomenon, esophageal disease, sclerodactyly, and telangiectases) and scleroderma. Generalized essential telangiectasia consists of extensive, sometimes symmetric, sheets of linear telangiectases, predominantly on the limbs or trunk. Sunlight and ionizing radiation may produce localized linear telangiectases in sun-exposed areas. Traumatic lesions are usually linear, or occasionally spider-like and localized. The telangiectases of acne rosacea are linear, limited to the face and nose, and spare mucous membranes. Venous lakes are deep blue, soft papules and nodules occurring on the lips and ears that blanch only partially on diascopy, are usually few in number, and are not associated with mucosal lesions.
Treatment
Treatment should be directed at controlling complications of arteriovenous malformations before they become symptomatic. Epistaxis can be controlled with nasal packing or with electrocautery. Coagulation may only offer temporary relief. Laser therapy with a neodymium:YAG (Nd:YAG) laser is known to control bleeding. The placement of a split-thickness skin graft to protect fragile telangiectatic vessels has been successful in 25% to 64% of patients. However, telangiectatic vessels recur at the mucosal border of the graft. Prophylactic lubrication of the nasal mucosa may provide relief. There is a tendency to reduce epistaxis with estrogen therapy.
If the site can be identified, then the gastrointestinal bleeding responds to electrocautery or laser via endoscopy. If these treatments are unsuccessful, surgical resection of involved bowel segments may be necessary. Low-dose combinations of estrogen and progesterone have successfully treated severe blood loss from enteric telangiectases.
Significant arteriovenous fistulas in any location are usually controlled by resection. Embolization with both detachable balloons and coils may significantly reduce the morbidity in pulmonary arteriovenous malformations. Cerebral arteriovenous malformations have been treated with balloon and coil embolization, stereotactic surgery, or conventional neurosurgery to prevent disabling hemorrhage.
Polyposis Syndromes
There are two recognized groups of hereditary polyposis: adenomatous polyposis syndromes and hamartomatous polyposis syndromes. Adenomatous polyposis syndromes have proven malignant potential. Hamartomatous polyposis syndromes represent malformation of the connective tissue of the intestinal mucosa. Even though they are classified as hamartomas, they still possess some risk of cancer.
Adenomatous Polyposis Syndromes
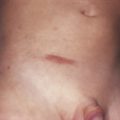
Familial adenomatous polyposis (FAP) refers to multiple adenomatous polyps numbering more than 100, and this condition predisposes to colorectal cancer with almost 100% certainty. FAP is characterized by autosomal dominant inheritance of colorectal polyposis without extracolonic manifestations. Considered to be a pathogenic variant of FAP, Gardner’s syndrome is characterized by an autosomal dominant inheritance with high penetrance and variable expressivity. Gardner’s syndrome entails colorectal polyposis with multiple epidermoid cysts ( Fig. 29-4 ), subcutaneous fibromas, lipomas, desmoid tumors, and osteomas of the facial bones and skull. If not treated, patients with FAP syndrome develop colon cancer beginning at ages of 20 to 30 years.