Lupus erythematosus (LE), dermatomyositis (DM), scleroderma, rheumatoid arthritis, Sjögren syndrome, eosinophilic fasciitis, relapsing polychondritis, and related disorders are classified as connective tissue diseases. Basic to all these is a complex array of autoimmune responses that target or affect collagen or ground substance.
Lupus Erythematosus
Lupus may manifest as a systemic disease or in purely cutaneous forms. Cutaneous manifestations of LE are classified as in Box 8.1 .
I
Chronic Cutaneous LE
- A.
Discoid LE
- 1.
Localized
- 2.
Disseminated
- 1.
- B.
Verrucous (hypertrophic) LE (Behçet): usually acral and often lichenoid
- C.
Lupus erythematosus–lichen planus overlap
- D.
Chilblain LE
- E.
Tumid lupus
- F.
Lupus panniculitis (LE profundus)
- 1.
With no other involvement
- 2.
With overlying discoid LE
- 3.
With systemic LE
- 1.
II
Subacute Cutaneous LE
- A.
Papulosquamous
- B.
Annular
- C.
Syndromes commonly exhibiting similar morphology
- 1.
Neonatal LE
- 2.
Complement deficiency syndromes
- 3.
Drug induced
- 1.
III. Acute Cutaneous LE: Localized or Generalized Erythema or Bullae, Generally Associated With SLE
LE, Lupus erythematosus; SLE, systemic lupus erythematosus.
Chronic Cutaneous Lupus Erythematosus
Discoid Lupus Erythematosus
Discoid lupus erythematosus (DLE) generally occurs in young adults, with women outnumbering men 2 : 1. Lesions begin as dull-red macules or indurated plaques that develop an adherent scale, then evolve with atrophy, scarring, and pigment changes ( Fig. 8.1 ). In darker-skinned individuals, lesions typically demonstrate areas of both hyperpigmentation and depigmentation. In lighter-skinned patients, the plaques may appear gray or have minimal pigment alteration. The hyperkeratosis characteristically extends into patulous follicles, producing carpet tacklike spines on the undersurface of the scale.
Localized Discoid Lupus Erythematosus.
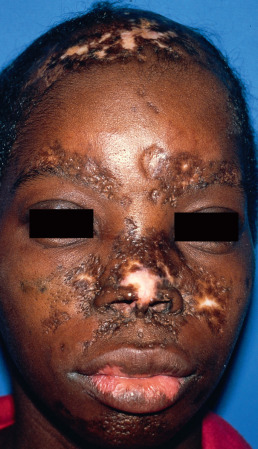
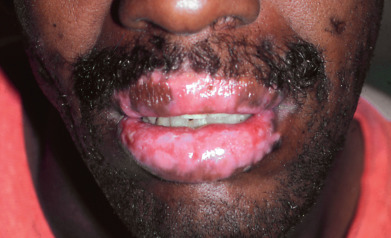
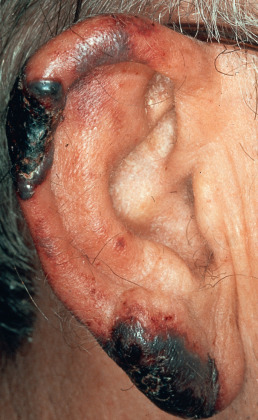
Discoid lesions are usually localized above the neck, particularly on sun-exposed sites such as the scalp, bridge of the nose, malar areas, lower lip, and ears ( Fig. 8.2 ). The concha of the ear and external canal are frequently involved. Typical lesions are disk-shaped areas of erythema, with follicular plugging, which progress to develop atrophy, scarring, and pigment alteration. Some patients present with periorbital edema and erythema. On the scalp, most lesions begin as erythematous patches or plaques that evolve into white, often depressed, hairless patches. Perifollicular erythema and the presence of easily extractable anagen hairs are signs of active disease. Scarred areas may appear completely smooth or may demonstrate dilated follicular openings in the few remaining follicles. Itching and tenderness are common but rarely severe. On the lips, lesions may be gray or red and hyperkeratotic. They may be eroded and are usually surrounded by a narrow, red inflammatory zone ( Fig. 8.3 ). In one study, 24% of DLE patients had mucosal involvement of the mouth, nose, eye, or vulva. Rarely, aggressive squamous cell carcinoma (SCC) may arise in long-standing lesions of DLE.
Patients with cutaneous lupus erythematosus (CLE) may develop systemic disease over time, sometimes with a lengthy delay (8–10 years), though overall only a small percentage (5%–23% reported, though more often the number is less than 15%) of patients with localized DLE will go on to meet criteria for systemic lupus erythematosus (SLE), usually due to cutaneous and mucosal criteria. Although progression from purely cutaneous DLE to SLE occurs infrequently, patients with SLE frequently have discoid lesions. These patients generally have systemic involvement early in the course of their disease, rather than evolving from chronic cutaneous LE to SLE. Fever and arthralgia are common in patients with SLE and discoid lesions. In patients with systemic symptoms, abnormal laboratory tests, such as elevation of antinuclear antibodies (ANAs), antibodies to double-stranded (ds) DNA and C1q, leukopenia, hematuria, and proteinuria, help to identify patients with SLE and suggest a prognosis.
Generalized Discoid Lupus Erythematosus.
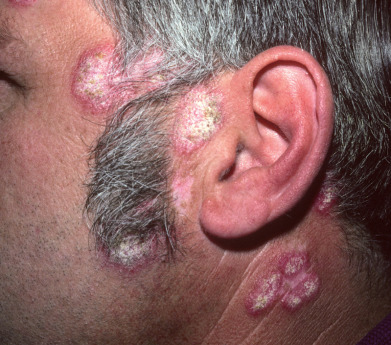
Generalized DLE is less common than localized DLE. All degrees of severity are encountered. Most often, the thorax and upper extremities are affected, as well as the head and neck ( Fig. 8.4 ). The scalp may become quite bald with striking patterns of hyperpigmentation and depigmentation. Diffuse scarring may involve the face and upper extremities. Laboratory abnormalities, such as an elevated erythrocyte sedimentation rate (ESR), elevated ANAs, single-stranded (ss) DNA antibodies, and leukopenia, are more common with this form of LE than with localized DLE, and as such patients with generalized DLE are more likely to meet criteria for systemic lupus.
Childhood Discoid Lupus Erythematosus.
Among children with DLE, a low frequency of photosensitivity and a higher rate of association with SLE have been noted, with one recent study showing 15% of patients with DLE had concurrent SLE, and 26% of the remaining patients eventually met SLE criteria. In most other respects, the clinical presentation and course are similar to those in adults.
Histology.
The epidermis may demonstrate effacement of the rete ridge pattern or irregular acanthosis. Compact hyperkeratosis without parakeratosis is characteristic, and follicular plugging is typically prominent. Hydropic degeneration of the basal layer of the epidermis and follicular epithelium results in pigmentary incontinence. A patchy perivascular and periadnexal lymphoid inflammatory infiltrate occurs in the superficial and deep dermis. The infiltrate characteristically surrounds vessels, follicles, and the eccrine coil. Increased mucin is often present, though is not a reliable mechanism to distinguish lupus from mimickers. Thickening of the basement membrane zone (BMZ) may be prominent.
The histology varies with the stage of the lesion. Acute lesions show only patchy lymphoid inflammation and vacuolar interface dermatitis. Lesions established for several months begin to show hyperkeratosis, BMZ thickening, and dermal mucin. Chronic, inactive lesions show atrophy, with postinflammatory pigmentation and scarring throughout the dermis. At this stage, the inflammatory infiltrate is sparse to absent. Pilosebaceous units, except for “orphaned” arrector muscles, are destroyed. At this stage, the dermis appears fibrotic, but an elastic tissue stain can still distinguish the diffuse dermal scar of lupus from the focal, wedge-shaped, superficial scars of lichen planopilaris (LPP) or folliculitis decalvans. Direct immunofluorescence (DIF) testing of lesional skin is positive in more than 75% of cases, provided the lesions have been active for at least several months and usually demonstrate strong, continuous granular deposition of immunoglobulin and complement located at the dermoepidermal junction (DEJ).
Differential Diagnosis.
Discoid LE must often be differentiated from seborrheic dermatitis, rosacea, lupus vulgaris, sarcoidosis, drug eruptions, actinic keratosis, Bowen disease, lichen planus (LP), tertiary syphilis, and polymorphous light eruption (PMLE). Seborrheic dermatitis does not show atrophy, alopecia, or dilated follicles and has greasy, yellowish scale without follicular plugs. Acral, lip, and scalp lesions of chronic cutaneous LE may demonstrate lichenoid dermatitis histologically. In these cases, the presence of continuous granular immunoglobulin in addition to cytoid bodies is a helpful distinguishing feature.
In rosacea, atrophy does not occur, and pustules are almost always found. Apple-jelly nodules (granulomas) are seen with diascopy in lupus vulgaris. Sunlight-sensitizing agents, such as sulfonamides, may produce lesions similar to LE, because phototoxic reactions demonstrate vacuolar interface dermatitis. It may be necessary to differentiate syphilis and sarcoid by biopsy and serologic testing. PMLE is distinguished by the absence of scarring and the presence of intensely edematous plaques and papules. DIF is generally negative or nonspecific in PMLE.
Hypertrophic Lupus Erythematosus
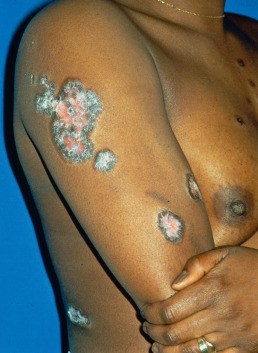
Nonpruritic papulonodular lesions may occur on the arms and hands, resembling keratoacanthoma or hypertrophic LP ( Fig. 8.5 ). The lips and scalp may also demonstrate lesions that resemble LP or LPP. Dermoscopy may help distinguish hypertrophic LE from SCC, but biopsy is often needed. Histologic sections of these lesions typically demonstrate lichenoid dermatitis, and a careful examination for other characteristic skin lesions of LE or LP, as well as DIF testing, may be critical in establishing a diagnosis. BMZ thickening, dermal mucin, eccrine coil involvement, and subcutaneous nodular lymphoid infiltrates are features of LE that are not found in LP. Recent reports suggesting the pattern and number of plasmacytoid dendritic cells may be helpful in distinguishing SCC from hypertrophic LE.
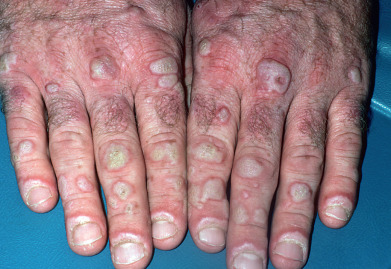
Lupus Erythematosus–Lichen Planus Overlap Syndrome
In addition to the cases of hypertrophic LE with lichenoid histology previously discussed, there are patients with a true overlap syndrome with features of both LE and LP. The lesions are usually large, atrophic, hypopigmented, red or pink patches and plaques. Pigment abnormalities become prominent over time, and fine telangiectasia and scaling are usually present. The extensor aspects of the extremities and midline back are typically affected. Prominent palmoplantar involvement is characteristic and tends to be the most troublesome feature for these patients. Nail dystrophy and anonychia may occur. Scarring alopecia and oral involvement have been noted in some patients. The histology of individual lesions has features of LP and/or LE. One study suggested that higher CD3 and CD34 may favor LP over DLE, but this has not been widely examined. DIF may help differentiate the two, but they may share overlapping features. Response to treatment is poor, although potent topical corticosteroids, dapsone, thalidomide, or isotretinoin may be effective. Some patients require immunosuppressive therapy with agents such as mycophenolate mofetil (MMF) or azathioprine. It should also be noted that antimalarials can occasionally produce a lichenoid drug eruption. A rare variant of chronic CLE termed the pigmented macular variant has been described, with lichenoid to gray pigmentation in sun exposed areas; although these patients display interface reaction on biopsy and may have seropositivity, systemic disease is rare, and it is unclear if this represents a rare subtype of lupus or another lichenoid entity. Patients respond to topical steroids, sun protection, and hydroquinone.
Chilblain Lupus Erythematosus
Chilblain LE (Hutchinson) is a chronic, unremitting form of LE affecting the fingertips, rims of ears, calves, and heels, especially in women. It is usually preceded by DLE on the face. Systemic involvement is sometimes seen. Mimicry of sarcoidosis may be striking. Cryoglobulins and antiphospholipid antibody (APLA) should also be sought. Familial chilblain lupus has been linked to mutations in a number of genes, particularly TREX-1, and is considered part of the group of genetic diseases termed interferonopathies.
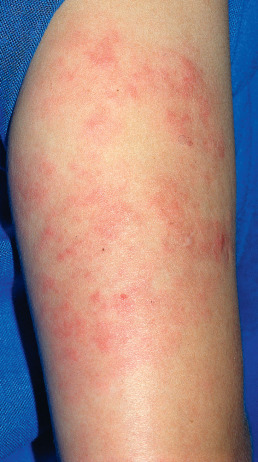
Tumid Lupus Erythematosus
Tumid LE is a rare but distinctive entity. Patients present with edematous erythematous plaques, usually on the trunk ( Fig. 8.6 ). Histologically, the lesions demonstrate a patchy superficial and deep perivascular and periadnexal lymphoid infiltrate that frequently affects the eccrine coil. Dermal mucin deposition is typical and may be striking. The lesions generally respond readily to antimalarials. Tumid LE shares many features with reticular erythematous mucinosis, and some authorities consider them to be closely related entities.
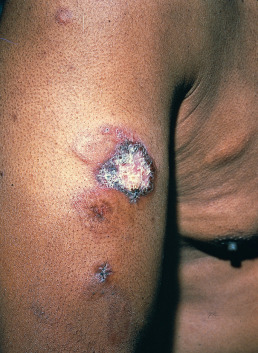
Lupus Erythematosus Panniculitis (Lupus Erythematosus Profundus)
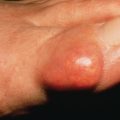
Patients with the panniculitis type of LE develop subcutaneous nodules that are usually firm, sharply defined, and nontender. The proximal extremities are typically involved. Usually, the overlying skin is normal, but overlying discoid or tumid lesions may occur ( Fig. 8.7 ). Some cases are discovered incidentally when an unrelated lesion is biopsied. The lesions may progress to deep depressions, or “dells,” from loss of the panniculus. LE panniculitis is characteristically chronic and occurs most often in women between ages 20 and 45. Many patients have DLE at other sites or less typically, in the overlying skin.
Histologic sections demonstrate lymphoid nodules in the subcutaneous septa, necrosis of the fat lobule, and fibrinoid or hyaline degeneration of the remaining lipocytes. Lipomembranous change, resembling frost on a windowpane, is more typical of stasis panniculitis (lipodermatosclerosis), but it may be noted focally in LE panniculitis. The overlying epidermis may show basal liquefaction and follicular plugging or may be normal. Dermal lymphoid nodules or vertical columns of lymphoid cells may be seen in fibrous tract remnants. Dermal mucin may be prominent, and dermal collagen hyalinization (resembling that seen in morphea) may be present. Continuous granular deposition of immunoglobulin and C3 may be seen at the DEJ. In active cases, abundant fibrin is usually noted in the panniculus.
The most important entity to consider in the differential diagnosis is subcutaneous panniculitis–like T-cell lymphoma (SPTCL). Important clues include the presence of lipocytes, rimmed by atypical lymphocytes with nuclear molding, and the presence of constitutional symptoms. Erythrophagocytosis may be present focally, and T-cell clonality can usually be demonstrated. The infiltrate may be CD8 dominant or may label strongly for CD56, as in natural killer cell lymphoma, or CD30, as in anaplastic lymphoma. CD5 and CD7 expression may be reduced (aberrant loss of pan-T markers). Unfortunately, T-cell clonality, erythrophagocytosis, CD8 predominance, and loss of CD5 or CD7 may also be seen in patients with LE panniculitis who respond to antimalarials or corticosteroids and do not progress to clinical lymphoma. Taken together, these data suggest that some cases of lymphoma may be virtually indistinguishable from LE panniculitis, or that some cases of LE panniculitis represent an abortive lymphoid dyscrasia. Patients diagnosed with lupus panniculitis should be monitored for progression.
Subacute Cutaneous Lupus Erythematosus
In 1979 Sontheimer, Thomas, and Gilliam described a clinically distinct subset of cases of LE to which they gave the name subacute cutaneous lupus erythematosus (SCLE). Patients are most often white women age 15–40. SCLE patients make up approximately 10%–15% of the LE population. Lesions are scaly and evolve as polycyclic annular lesions ( Fig. 8.8 ) or psoriasiform plaques, although rare widespread cases of erythrodermic SCLE or patients resembling SJS/TEN have been described. The lesions vary from red to pink with faint violet tones. The scale is thin and easily detached, and telangiectasia or dyspigmentation may be present. Follicles are not involved, the lesions tend to be transient or migratory, and there is no scarring. Lesions tend to occur on sun-exposed surfaces of the face and neck, the V portion of the chest and back, and the sun-exposed areas of the arms. Photosensitivity is prominent in about half of patients. Concomitant DLE is present in 20% of cases.

About three quarters of patients have arthralgia or arthritis, 20% have leukopenia, and 80% have a positive ANA test (usually in a particulate pattern). Between 20% and 50% of patients meet the American Rheumatology Association (ARA) criteria for a diagnosis of SLE. The majority of cases have antibodies to Ro/SSA antigen, and most are positive for human leukocyte antigen (HLA) DR3. La/SSB may also be present, and many patients have overlap features with Sjögren syndrome. The disease generally runs a mild course, and renal, central nervous system (CNS), or vascular complications are unusual. An association with autoimmune thyroid disease has been noted. Most patients respond to sun protection and antimalarial agents. From 12%–30% of cases may be drug induced. Medication-induced SCLE is traditionally most often related to hydrochlorothiazide due to its wide use, but proton pump inhibitors are increasingly recognized, perhaps due to over-the-counter availability. Drug-induced SCLE may also be seen with angiotensin-converting enzyme (ACE) inhibitors, calcium channel blockers (CCBs), interferons (IFNs), anticonvulsants, griseofulvin, glyburide, piroxicam, penicillamine, spironolactone, terbinafine, statins, voriconazole, and chemotherapeutic agents. Chemotherapeutic agents implicated include fluorouracil, capecitabine, cemcitabine, docetaxel, paclitaxel, and doxorubicin, with emerging reports of nivolumab, other checkpoint inhibitors, and small molecule kinase inhibitors such as masitinib. TNF inhibitors are increasingly recognized as the triggering drug and are important to keep in mind due to the potential for psoriasiform SCLE to resemble psoriasis, which can lead to a misdiagnosis of TNF-induced psoriasis—photosensitivity is a clue to SCLE in these cases. Case reports describe both topical terbinafine and imiquimod inducing SCLE-like eruptions as well.
Histopathology
Vacuolar interface dermatitis is a universal finding in active SCLE lesions. Mild hyperkeratosis and parakeratosis may be present. Chronic changes of DLE, such as follicular plugging, BMZ thickening, and heavy lymphoid aggregates, are usually lacking. Dermal mucin is variable. DIF is positive in lesional skin in only about one third of cases. A dustlike particulate deposition of immunoglobulin G (IgG) in epidermal nuclei of Ro-positive patients may be present and is a helpful diagnostic finding.
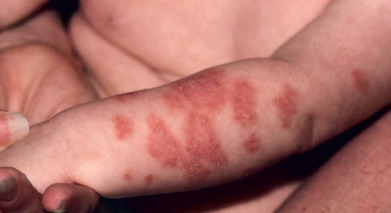
Neonatal Lupus Erythematosus
Most infants with neonatal lupus are girls, born to mothers who carry the Ro/SSA antibody. These infants have no skin lesions at birth, but develop them during the first few weeks of life. Annular erythematous macules and plaques may appear on the head and extremities ( Fig. 8.9 ). Periocular involvement (raccoon eyes) may be prominent. With time, the lesions fade and become atrophic. Telangiectasia or dermal mucinosis in an acral papular pattern may be the predominant findings in some cases. Telangiectatic macules or angiomatous papules may be found in sun-protected sites such as the diaper area, may occur independently of active lupus skin lesions, and may be persistent. The skin lesions usually resolve spontaneously by 6 months of age, and usually heal without significant scarring, although atrophy and telangiectatic mats may persist. Dyspigmentation and persistent telangiectasias may remain for months to years. Half the mothers are asymptomatic at delivery, although many will subsequently develop arthralgia, Sjögren syndrome, or other mild systemic findings.
Although the skin lesions are transient, half the patients have an associated isolated congenital heart block, usually third degree, which is permanent. Some infants have only this manifestation of LE, and for cardiac lesions alone, there is no female predominance. In children with cutaneous involvement, thrombocytopenia and hepatic disease may occur as frequently as cardiac disease. Close monitoring is recommended for at least the first 3 months of life, and by 9 months of life most cases resolve, with only 10% of infants with persistent antibodies at that time.
There is a strong association with Ro/SSA autoantibody. Almost all mothers, and thus almost all infants, are positive for this antibody, although some mothers are also positive for La/SSB, and some with only U1RNP antibodies have been described. Infants with only U1RNP antibodies have not developed heart block. There is linkage to HLA-DR3 in the mother. The risk that a second child will have neonatal LE is approximately 25%. Japanese infants apparently differ in that they may express anti-dsDNA antibodies, and 8% progress to SLE. In unselected women with anti-Ro antibodies, only 1%–2% will have an infant with neonatal LE.
Complement Deficiency Syndromes
Although deficiency of many complement components may be associated with LE-like conditions, deficiencies of the early components, especially C2 and C4, are most characteristic. Many such cases are found to have photosensitive annular SCLE lesions and Ro/SSA antibody formation. Patients with C4 deficiency often have hyperkeratosis of the palms and soles. Heterozygous deficiency of either complement component C4A or C4B has a frequency of approximately 20% in white populations. Homozygous deficiency of both is rare, and affected patients may present with SLE with mesangial glomerulonephritis, membranous nephropathy, and severe skin lesions. Although frequently asymptomatic, homozygous C2 deficiency can cause severe infections, SLE, and atherosclerosis.
Systemic Lupus Erythematosus
Young to middle-aged women are predominantly affected by SLE, manifesting a wide range of symptoms and signs. Skin involvement occurs in 80% of cases and is often helpful in arriving at a diagnosis. Its importance is suggested by the fact that 4 of the 11 American College of Rheumatology (ACR) criteria for the diagnosis of SLE are mucocutaneous findings. The diagnostic criteria are as follows:
- 1.
Malar rash
- 2.
Discoid rash
- 3.
Photosensitivity
- 4.
Oral ulcers (21%)
- 5.
Arthritis
- 6.
Proteinuria >0.5 g/day or casts
- 7.
Neurologic disorders (seizures or psychosis in the absence of other known causes)
- 8.
Pleuritis/pericarditis
- 9.
Blood abnormalities (e.g., hemolytic anemia, leukopenia, thrombocytopenia)
- 10.
Immunologic disorders, including anti-dsDNA antibody, anti-Sm, APLAs (based on IgG or IgM anticardiolipin antibodies, lupus anticoagulant [LA], or false-positive serologic test for syphilis known for at least 6 months)
- 11.
Positive ANA blood test
For identification of patients in clinical studies, a patient may be said to have SLE if four or more of these criteria are satisfied, serially or simultaneously. It is important to note that many patients present with autoantibodies, arthralgia, and constitutional signs, but do not meet ACR criteria for SLE. With time, patients may evolve to meet all criteria. The Systemic Lupus International Collaborating Clinics (SLICC) group revision of the ACR criteria results in greater sensitivity with equal specificity. According to the SLICC rule, the patient must manifest at least four criteria (including at least one clinical criterion and one immunologic criterion) or must have biopsy-proven lupus nephritis in the presence of either ANAs or anti-dsDNA antibodies. Many patients with DLE or SCLE meet criteria for SLE due to mucocutaneous criteria.
Cutaneous Manifestations
The characteristic butterfly facial erythema seen in patients with SLE is a common manifestation of acute cutaneous LE. The eruption usually begins on the malar area and the bridge of the nose. There may be associated edema. The nasolabial crease if often strikingly spared, unlike in DM. The ears and chest may also be the sites of early lesions. Biopsies at all sites show interface dermatitis and a scant perivascular lymphoid infiltrate. The eruption may last a day to several weeks and resolves without scarring. There may be more widespread erythema in some cases.
Bullous lesions of lupus erythematosus (BLE) occur as single or grouped vesicles or bullae, often widespread, with a predilection for sun-exposed areas ( Fig. 8.10 ). Rarely, the lesions may itch. Most sets of published criteria require that patients with BLE meet ACR criteria for SLE, but some patients have identical bullous lesions and less than four ACR criteria. Histologically, neutrophils accumulate at the DEJ and within dermal papillae. In bullous lesions, there is a subepidermal bulla or superficial dermal edema containing neutrophils. Fluorescence with IgG, IgM, IgA, or C3 is typically present in a continuous, granular pattern at the BMZ on DIF testing. Neutrophils are found in or below the lamina densa on immunofluorescent electron microscopy. Most of these patients are HLA-DR2 positive. The recognition of this subset as distinct is made clear by its often dramatic therapeutic response to dapsone. Epidermolysis bullosa acquisita is histopathologically and immunopathologically identical, because both diseases are mediated by circulating antibodies against type VII collagen. Dapsone is usually ineffective in epidermolysis bullosa acquisita.
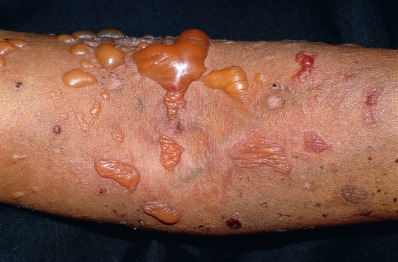
A variety of vascular lesions occur in 50% of SLE patients. Often, fingertips or toes show edema, erythema, or telangiectasia. Nailfold capillary loops in LE are more likely to show wandering glomeruloid loops, whereas DM and scleroderma capillary loops demonstrate symmetric dilation and dropout of vessels. Capillary loops in the Osler-Weber-Rendu syndrome demonstrate ectasia of half the capillary loop. Erythema multiforme (EM)–like lesions may predominate, termed Rowell syndrome. Rarely, lupus may present with TEN-like lesions due to intense interface inflammation leading to blistering.
In addition to periungual telangiectasia, red or spotted lunulae may be present in patients with SLE, as in RA. The palms, soles, elbows, knees, or buttocks may become persistently erythematous or purplish, sometimes with overlying scale. Diffuse, nonscarring hair loss is common. Short hairs in the frontal region are called “lupus hairs.” These hairs result from a combination of chronic telogen effluvium and increased hair fragility.
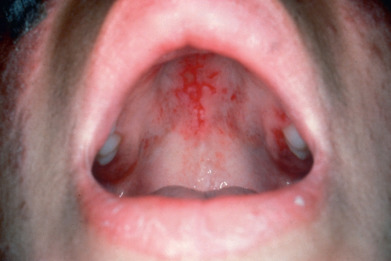
Mucous membrane lesions are seen in 20%–30% of SLE patients. Conjunctivitis, episcleritis, and nasal and vaginal ulcerations may occur. Oral mucosal hemorrhages, erosions, shallow angular ulcerations with surrounding erythema, and gingivitis are common ( Fig. 8.11 ). Erythema, petechiae, and ulcerations may occur on the hard palate.
Multiple eruptive dermatofibromas have been described in SLE. Leg ulcers, typically deeply punched out and with very little inflammation, may be seen on the pretibial or malleolar areas. Many of these patients present with a livedoid pattern, and many have an antiphospholipid antibody. Sneddon syndrome is composed of livedo reticularis and strokes related to a hyalinizing vasculopathy.
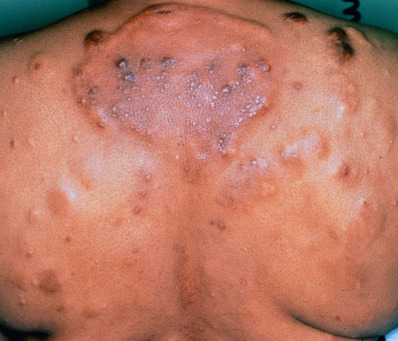
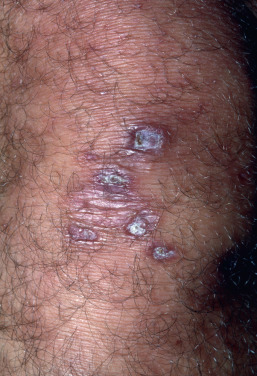
Calcinosis cutis is uncommon but may be dramatic. Also seen infrequently are plaquelike or papulonodular depositions of mucin. These reddish purple to skin-colored lesions are often present on the trunk and arms or head and neck ( Fig. 8.12 ). Last, a symmetric papular eruption of the extremities may occur. These skin-colored to erythematous lesions with a smooth, ulcerated or umbilicated surface may show vasculitis or, in older lesions, a palisaded granulomatous inflammation. These occur in patients with SLE, RA, or other immune complex–mediated disease. This eruption has been referred to as palisaded neutrophilic and granulomatous dermatitis of immune complex disease ( Fig. 8.13 ).
Systemic Manifestations
Most organs can be involved in SLE; the symptoms and findings are often caused by immune complex disease, especially vasculitis. The earliest changes noted may be transitory or migratory arthralgia, often with periarticular inflammation. Fever, weight loss, pleuritis, adenopathy, or acute abdominal pain may occur. Arthralgia is often the earliest abnormality and may remain the sole symptom for some time. About 95% of SLE patients will manifest this symptom. Arthralgia, deforming arthropathy, and acute migratory polyarthritis resembling RA may all occur as manifestations of SLE. Avascular necrosis of the femoral head has been observed. Although this is a known complication of systemic corticosteroid therapy, it has also occurred in patients with SLE who have never taken corticosteroids.
Patients with SLE have a higher rate of peripheral arterial disease compared with controls. Thrombosis in vessels of various sizes and thromboembolism may be a recurring event ( Fig. 8.14 ). It may be attributed to a plasma constituent, paradoxically called “lupus anticoagulant” (LA) because it causes prolonged coagulation studies in vitro but thrombosis in vivo. The finding of an LA is usually associated with APLAs. These may be anticardiolipin antibodies, but other APLA types—antiphosphatidylserine, antiphosphatidylinositol, and antiphosphatidylethanolamine—may occur. APLAs and elevated homocysteine may each increase the risk of thrombosis. APLAs are associated with early-onset organ damage. SLE patients with APLAs have higher mortality. Many, but not all, patients have a false-positive blood test for syphilis. In one study, inflammatory lesions of SLE and infections were the most common causes of death during the initial 5 years of disease, whereas thromboses were the most common cause of death after the first 5 years.
Renal involvement is a predictor of poor prognosis. It may be of either nephritic or nephrotic type, leading in either case to chronic renal insufficiency with proteinuria and azotemia. Patients with lupus who are ANCA positive have higher rates of damage. Active nephritis is unlikely in the absence of anti-dsDNA. Both anti-dsDNA antibody and anti-C1q antibody are of relatively high specificity for active nephritis. Hypercholesterolemia and hypoalbuminemia may occur. Immunoglobulin and complement components have been found localized to the BMZ of glomeruli, where vasculitis produces the characteristic “wire-loop” lesion.
Myocarditis is indicated by cardiomegaly and gallop rhythm, but the electrocardiographic (ECG) changes are usually not specific. Pericarditis, the most frequent cardiac manifestation, and endocarditis also occur. Patients with SLE have accelerated atherosclerosis and elevated risk for cardiovascular morbidity and mortality. Raynaud phenomenon (RP) occurs in about 15% of patients, who have less renal disease and consequently lower mortality rates.
The CNS may be involved with vasculitis, manifested by hemiparesis, convulsions, epilepsy, diplopia, retinitis, and choroiditis. Livedo reticularis is a marker for patients at risk for CNS lesions (Sneddon syndrome; see earlier). Depression and anxiety are common in patients with SLE, and other psychiatric and personality have been reported as well.
Idiopathic thrombocytopenic purpura is occasionally the forerunner of SLE. Coombs-positive hemolytic anemia, neutropenia, and lymphopenia are other hematologic findings. The severity of thrombocytopenia may be a prognostic marker, but can also be followed as a biomarker for response to treatment. Gastrointestinal (GI) involvement may produce symptoms of nausea, vomiting, and diarrhea, and patients may develop intestinal pseudo obstruction. Frequently, the intestinal wall and the mesenteric vessels show vasculitis. Pulmonary involvement with pleural effusions, interstitial lung disease, pulmonary artery hypertension (PAH), and acute lupus pneumonitis may be present. Patients with SLE, pericardial effusions, and anti-RNP antibodies have higher rates of PAH. Sjögren syndrome (keratoconjunctivitis sicca) and Hashimoto thyroiditis are associated with SLE. Overlap with any of the connective tissue diseases may be seen, occurring in approximately 25% of patients. Muscular atrophy may accompany extreme weakness so that DM may be suspected. Myopathy of the vacuolar type may produce muscular weakness, myocardial disease, dysphagia, and achalasia of the esophagus. Steroid myopathy may also occur, and antimalarials may rarely induce myopathy. The serum aldolase level may be elevated with a normal creatine phosphokinase.
A history of exposure to excessive sunlight before the onset of the disease or before an exacerbation is sometimes obtained. Some patients may have only mild constitutional symptoms for weeks or months, but immediately after exposure to strong sunlight, they may develop the facial eruption and severe disease complications.
Hydralazine, procainamide, sulfonamides, penicillin, anticonvulsants, minocycline, and isoniazid have been implicated as causes of drug-induced LE. Penicillamine induces (or unmasks) true SLE. HLA-DR4 individuals, who are slow acetylators, are predisposed to develop hydralazine-induced LE. Antibody to the histone complex H2A-H2B is closely associated with disease. Most drug-induced SLE is associated with a positive ANA, and antibodies directed against histones. Exceptions include penicillamine and etanercept, which may induce or unmask native disease with anti-dsDNA antibodies. l -Canavanine, an amino acid found in alfalfa sprouts and tablets, can also induce or worsen SLE. TNF-inhibitors can induce a variety of types of lupus, with infliximab surpassing etanercept as the most commonly reported culprit. Patients with anti–chimeric antibodies may be ANA positive, and patients with TNF-induced lupus frequently display a malar rash, photosensitivity, and meet ACR criteria for lupus.
Childhood Systemic Lupus Erythematosus
The onset of childhood SLE occurs between ages 3 and 15, with girls outnumbering boys 4 : 1. The skin manifestations may be the typical butterfly eruption on the face and photosensitivity. In addition, there may be morbilliform, bullous, purpuric, ulcerating, or nodose lesions. The oral mucosa is frequently involved. Skin eruptions may be associated with joint, renal, neurologic, and GI disease. Weight loss, fatigue, hepatosplenomegaly, lymphadenopathy, and fever are other manifestations. Pediatric patients with SLE and APLAs, specifically lupus anticoagulants, are at high risk of developing thromboembolic events.
Pregnancy and Female Hormones
Women with LE may have successful pregnancies, but they might have difficulty conceiving, and miscarriages occur with greater frequency, especially among those with APLAs. The course of pregnancy may be entirely normal, with remission of the LE, or the symptoms of LE may become worse. Risk of fetal death is increased in women with a previous history of fetal loss and anticardiolipin or anti-Ro antibodies. For the patient with these antibodies but without a history of previous fetal loss, the risk of fetal loss or neonatal lupus is low. In most cases, the pregnancy itself is well tolerated, although a flare of SLE may occur during the postpartum period. Although several studies failed to demonstrate a clinically significant association between oral contraceptive (OC) use and flares of SLE, recently a UK epidemiologic study found an increased risk of SLE with current use of OC, and the U.S. Nurses’ Health Study suggested that OC and HRT were associated with SLE. There is a high incidence of thromboses in women with APLAs, and OCs containing second- or third-generation progestogens may induce a higher risk. One female-to-male transgender patient experienced significant improvement after testosterone therapy.
Etiology
Genetics plays an important role in LE, with over 100 loci identified in association with sLE. Many of these gene targets are involved in recognition of nucleic acid, type 1 IFN production, and immune signaling. Taken together, these data suggest polygenetic susceptibility to LE. A family history of connective tissue disease is a strong risk factor for all forms of LE. Monozygotic twins, however, display a low concordance rate, with lupus in approximately one fourth of cases. Some skin lesions of LE follow lines of Blaschko, suggesting postzygotic mutation or loss of heterozygosity for a genetic locus. Understanding of lupus genetics is rapidly evolving.
The C-reactive protein (CRP) response is defective in patients with flares of SLE, and the gene locus for CRP maps to 1q23.2 within an interval linked with SLE. Gene polymorphisms in APRIL, a member of the TNF family, have also been linked with SLE. Increased expression of TNF-α and IFN-inducible proteins are noted in cutaneous LE. Polymorphisms of the C1qA gene are associated with both systemic and cutaneous LE. HLA-DR3 confers risk for SLE, and C4 null alleles in Europeans is a specific genetic risk factor for SLE. Linkage varies in different ethnic groups and different clinical subsets of lupus.
Whole exome sequencing has helped identify rare monogenic variants of SLE and SLE-like diseases. CYBB mutations have an X-linked inheritance, and carriers have a higher risk of DLE and SLE. Mutations in DNASE1L3 are associated with SLE with early onset and anti-dsDNA abs. TREX1 mutations are associated with familial chilblain lupus.
Several aspects of the altered immune response are worth particular attention. T-suppressor cell function is reduced in patients with LE. Overproduction of γ-globulins by B cells and reduced clearance of immune complexes by the reticuloendothelial system may contribute to complement-mediated damage. Externalization of cellular antigens, such as Ro/SSA in response to sunlight, may lead to cell injury by way of antibody-dependent cellular cytotoxicity. Abnormal apoptosis or reduced clearance of apoptotic cells may lead to increased exposure of nucleosome antigens and antinucleosome antibodies.
Both ultraviolet (UV) B and UVA can upregulate antigen expression and cytokines, including IL-1, TNF-α, and LFA-1, causing release of sequestered antigens and free radical damage, with UV B felt to be the most pathologically relevant. All these mechanisms may contribute to photosensitivity and UV-induced flares of systemic disease. Air pollutants have been considered a potential trigger, due to increased concentrations of SLE in urban areas and a small Canadian study suggesting a possible link. Tobacco smoke is highly associated with cutaneous LE, but not with systemic LE, and may interfere with the efficacy of antimalarial therapy. Minimal credible data exist regarding other possible aggravating dietary factors, but some reports have implicated excess calories, excess protein, high fat (especially saturated and ω-6 polyunsaturated fatty acids), excess zinc, and excess iron.
Laboratory Findings
There may be hemolytic anemia, thrombocytopenia, lymphopenia, or leukopenia; ESR usually is greatly elevated during active disease but nonspecific. Coombs test may be positive, there is a biologic false-positive test for syphilis, and a rheumatoid factor (RF) may be present. IgG levels may be high, the albumin/globulin ratio is reversed, and serum globulin is increased, especially the γ-globulin or α2 fraction. Albumin, red blood cells, and casts are the most frequent findings in the urine.
Immunologic Findings
- 1.
ANA test. This is positive in 95% of cases of SLE. Human substrates, such as Hep-2 or KB tumor cell lines, are far more sensitive than mouse substrates, and the historical entity “ANA-negative lupus” is quite rare now. ANA pattern has some correlation with clinical subsets, such as a shrunken peripheral pattern in SLE with renal disease, a fine particulate pattern in subacute cutaneous LE, and a homogeneous pattern with antihistone antibodies.
- 2.
Double-stranded DNA. Anti-dsDNA, anti–native DNA. This is specific, but not very sensitive. It indicates a high risk of renal disease, and correlates with a shrunken peripheral ANA pattern and positive DIF in sun-protected skin.
- 3.
Anti-Sm antibody. Sensitivity is less than 10%, but specificity is very high.
- 4.
Antinuclear ribonucleic acid protein (anti-nRNP). Very high titers are present in mixed connective tissue disease (MCTD). Lower titers may be seen in SLE.
- 5.
Anti-La antibodies. These are common in SCLE and Sjögren syndrome, and occasionally found in SLE.
- 6.
Anti-Ro antibodies. These are found in about 25% of SLE and 40% of Sjögren patients. They are more common in patients with SCLE (70%), neonatal LE (95%), C2- and C4-deficient LE (50%–75%), late-onset LE (75%), and Asian patients with LE (50%–60%). Photosensitivity may be striking, and externalization of the antigen is seen after UV exposure.
- 7.
Serum complement. Low levels indicate active disease, often with renal involvement.
- 8.
Lupus band test. Direct cutaneous immunofluorescence. Continuous granular deposits of immunoglobulins and complement along the DEJ occur in more than 75% of well-established lesions of DLE. In SLE, it usually is positive in sun-exposed skin. A positive test in normal, protected skin correlates with the presence of anti-dsDNA antibodies and renal disease. The lupus band test is seldom performed, because the same population of patients can be detected with anti-dsDNA antibodies.
- 9.
Anti-ssDNA antibody. This test is sensitive but not specific. Many patients are photosensitive. An IgM isotope seen in DLE may identify a subset of patients at risk for developing systemic symptoms.
- 10.
Antiphospholipid antibodies. Both the anticardiolipin antibody and the LA are subtypes of APLAs. These are associated with a syndrome that includes venous thrombosis, arterial thrombosis, spontaneous abortions, and thrombocytopenia. Livedo reticularis is a frequent skin finding, and nonfading acral microlivedo, with small, pink cyanotic lesions on the hands and feet, is a subtle clue to the presence of APLAs. These antibodies may occur in association with lupus and other connective tissue disease, or as a solitary event. In the latter case, it is referred to as the primary antiphospholipid syndrome.
Differential Diagnosis
Diagnostically, SLE must be differentiated from DM, EM, polyarteritis nodosa, acute rheumatic fever, RA, pellagra, pemphigus erythematosus (Senear-Usher syndrome), drug eruptions, hyperglobulinemic purpura, Sjögren syndrome, and myasthenia gravis. The SLE patient may have fever, arthralgia, weakness, lassitude, diagnostic skin lesions, increased ESR, cytopenias, proteinuria, immunoglobulin deposition at DEJ, and positive ANA test. Biopsies of skin lesions and involved kidney may also be diagnostic.
Treatment
Some general measures are important for all patients with LE. Exposure to sunlight must be avoided, and broad spectrum, high sun protection factor (SPF) sunscreen should be used daily. Photosensitivity is frequently present even if the patient denies it, and all patients must be educated about sun avoidance and sunscreen use. Smokers should be encouraged and assisted in quitting. The patient should also avoid exposure to excessive cold, to heat, and to localized trauma. Biopsies and scar revision will often provoke a flare of the disease. Women with SLE have an increased risk of osteoporosis, independent of corticosteroid use. Bone density should be monitored and calcium and vitamin D supplementation considered, especially with strict photoprotection. Vitamin D repletion was shown to improve response to some medications. Some women will benefit from bisphosphonate therapy, especially if corticosteroids are used. The most rapid bone loss with corticosteroid therapy occurs at the onset of treatment, so bisphosphonate therapy should not be delayed. Patients who will be treated with immunosuppressive agents should receive a tuberculin skin test, appropriate vaccinations, and thorough physical examination. Aggressive treatment is often necessary for discoid lesions and scarring alopecia. The slowly progressive nature of these lesions, and the lack of systemic involvement, may lead to inappropriate therapeutic complacency. The result is slow, progressive disfigurement.
Local Treatment.
The application of potent or superpotent topical corticosteroids is beneficial in LE patients. Occlusion may be necessary and may be enhanced by customized vinyl appliances (especially for oral lesions) or surgical dressings. Tape containing corticosteroid (Cordran) is sometimes helpful. The single most effective local treatment is the injection of corticosteroids into the lesions. Triamcinolone acetonide, 2.5–10 mg/mL, is injected at intervals of 4–6 weeks. No more than 40 mg of triamcinolone should be used at one time. Steroid atrophy is a valid concern, but so are the atrophy and scar produced by the disease. The minimal intralesional dose needed to control the disease should be used; when the response is poor, however, it is generally better to err on the slightly more aggressive side of treatment than to undertreat. Topical calcineurin inhibitors may also be useful as second-line topical therapy. Topical retinoids have scattered reports of benefits, particularly for hypertrophic lesions. Cryosurgery has been used but may induce scarring, dyspigmentation, and flared. Although lupus is a photosensitive disorder, UVA-I therapy appears to be a useful adjuvant treatment modality in some patients; all light should be used with caution. Photodynamic therapy has been reported as effective. Lasers such as the PDL 585–595 nm laser have been used to treat individual lesions with improvement, but patients should be treated cautiously at low fluence if attempted.
Systemic Treatment.
The safest class of systemic agent for LE is the antimalarials. Retinoids may be particularly helpful in treating hypertrophic LE. Systemic immunosuppressive agents are often required to manage recalcitrant cases or patients with systemic manifestations. Thalidomide can be effective, but its use is limited by the risk of teratogenicity and neuropathy. Methotrexate is often the next traditional systemic agent selected. Dapsone is the drug of choice for bullous systemic LE and may be effective in some cases of SCLE and DLE. Oral prednisone is generally reserved for acute flares of disease. Biologic agents and new targeted therapies are now used for refractory disease. One study of 73 antimalarial-refractory CLE patients showed that thalidomide was effective in 10 of 11, methotrexate in 10 of 19, dapsone in 8 of 18, belimumab in 6 of 16, MMF in 9 of 25, and azathioprine in 3 of 12.
Antimalarials.
Hydroxychloroquine (Plaquenil), at a dose of 5 mg/kg/day or less, has an excellent safety profile and is generally used as first-line systemic therapy in most forms of cutaneous LE, with two thirds of patients responding. If no response occurs after 3 months, another agent should be considered. Chloroquine (Aralen) is effective at 250 mg/day for an average adult but is difficult to procure and has higher rates of ocular toxicity. Quinacrine (Atabrine), 100 mg/day, may be added to hydroxychloroquine because it adds no increased risk of retinal toxicity. Quinacrine is also difficult to procure and carries a higher risk of yellowish pigmentation than the other antimalarials. Systemic treatment can sometimes be reduced or stopped during the winter months. A Cochrane group review of randomized controlled trials (RCTs) concluded that hydroxychloroquine and acitretin appear to be of similar efficacy, although adverse effects are more severe and occur more often with acitretin.
Ocular toxicity is rare with doses of hydroxychloroquine of 5 mg/kg/day or less, and tends to occur in older patients, patients with liver or kidney disease, or after 5 years of therapy. Ophthalmologic consultation should be obtained before, and approximately annually (depending on risk factors, this may not be necessary in the first few years of treatment). The finding of any visual field defect or pigmentary abnormality is an indication to stop antimalarial therapy.
Other reported side effects with antimalarials include hair loss, lichenoid eruptions, erythroderma, EM, purpura, urticaria, nervousness, tinnitus, abducens nerve paralysis, toxic psychoses, leukopenia, and thrombocytopenia. Antimalarials, except in very small doses, will exacerbate skin disease or cause hepatic necrosis in patients with porphyria cutanea tarda. They may also worsen or induce psoriasis. Quinacrine produces a yellow discoloration of the skin and conjunctivae. Antimalarials have also been known to produce blue-black pigmentation of the hard palate, nail beds, cartilage of the ears, alae nasi, and sclerae. Nausea, vomiting, anorexia, and diarrhea may develop. Aplastic anemia has rarely been noted in long-term therapy. A patient’s brown or red hair may rarely turn light blond. Morbilliform eruptions are rare in patients with lupus, but occur in a quarter of patients with DM.
Corticosteroids.
Systemic corticosteroids are highly effective for widespread or disfiguring lesions, but disease activity often rebounds quickly when the drug is discontinued. Because of long-term side effects, corticosteroid treatment should be limited to short courses to treat flares of disease or to obtain initial control while antimalarial therapy is being initiated. In patients with renal or neurologic involvement, corticosteroids should be administered in doses adequate to control the disease while treatment with a steroid-sparing regimen is initiated. Treatment with 1000 mg/day intravenous methylprednisolone for 3 days, followed by oral prednisone, 0.5–1 mg/kg/day, is effective in quickly reversing most clinical and serologic signs of activity of lupus nephritis. In general, the corticosteroid dose should be optimized to the lowest possible that controls symptoms and laboratory abnormalities.
Traditional Immunosuppressive Therapy.
Aggressive treatment protocols with agents such as pulse cyclophosphamide (with hydration and mesna to prevent bladder toxicity) have greatly improved the outcome of renal LE. Other immunosuppressive agents (e.g., azathioprine, methotrexate, MMF), are often employed as steroid-sparing agents for refractory cutaneous disease. Methotrexate is often effective; patients with concomitant renal disease may be at higher risk of severe methotrexate toxicity, and all patients should receive folic acid concurrently. Some authorities have suggested that azathioprine is inferior to MMF in the treatment of cutaneous lesions.
Other Therapy.
Isotretinoin therapy, 0.2–1 mg/kg/day, may be effective, especially in patients with hypertrophic or lichenoid lesions of LE. Rapid relapse may be noted when the drug is discontinued. Dapsone, clofazimine, acitretin, IFN alpha-2a, auranofin (oral gold), high-dose intravenous immune globulin (IVIG), and efalizumab have all been reported as effective in anecdotal use or limited trials. Thalidomide may be effective in antimalarial-refractory CLE, but is often limited by side effects including peripheral neuropathy. Lenalidomide may be more effective with lower rates of neurotoxicity and fewer side effects, and has increasingly been used to treat recalcitrant cases of CLE and SCLE, with response rates nearing 90%. Apremilast reduced disease activity in a pilot study of eight patients. Belimumab, a monoclonal antibody against B lymphocyte stimulator, was approved for SLE, but effects on cutaneous disease remain unclear. Anti-CD20 monoclonal antibody (rituximab) has been used successfully to treat life-threatening refractory SLE with renal and CNS involvement, as well as for hypocomplementemic urticarial vasculitis and refractory cutaneous lesions, though an RCT for SLE did not demonstrate success. Rituximab is also used for dapsone-resistant cases of BLE. Interleukin-6 (IL-6)–receptor inhibition with tocilizumab appears promising but may cause neutropenia. Sirukumab, another IL-6 antibody, was used in a study of 31 patients, with 91% experiencing adverse events and a nonsignificant decrease in disease activity.
Janus kinase (JAK) inhibitors are being explored for multiple interferonopathies and may have a role in treating lupus, and targeted anti-IFN antibodies are being evaluated as well. Ustekinumab has been reported as beneficial in a few cases of recalcitrant DLE.
Aggarwal N: Drug-induced subacute cutaneous lupus erythematosus associated with proton pump inhibitors. Drugs Real World Outcomes 2016; 3: 145.
Arkin LM, et al: The natural history of pediatric-onset discoid lupus erythematosus. J Am Acad Dermatol 2015; 72: 628.
Böckle BC, Sepp NT: Smoking is highly associated with discoid lupus erythematosus and lupus tumidus. Lupus 2015; 24: 669.
Chasset F, Arnaud L: Targeting interferons and their pathways in systemic lupus erythematosus. Autoimmun Rev 2018; 17: 44.
Costa-Reis P, et al: Monogenic lupus. Curr Opin Immunol 2017; 49: 87.
Deng Y, et al: Updates in lupus genetics. Curr Rheumatol Rep 2017; 19: 68.
Elman SA, et al: Development of classification criteria for discoid lupus. J Am Acad Dermatol 2017; 77: 261.
Ezra N, et al: Voriconazole-induced subacute cutaneous lupus erythematosus. Skinmed 2016; 14: 461.
Fennira F, et al: Lenalidomide for refractory chronic and subacute cutaneous lupus. J Am Acad Dermatol 2016; 74: 1248.
Fiehn C: Familial chilblain lupus. Curr Rheumatol Rep 2017; 19: 61.
Fruchter R, et al: Characteristics and alternative treatment outcomes of antimalarial-refractory cutaneous lupus erythematosus. JAMA Dermatol 2017; 153: 937.
Garza-Mayers AC, et al: Review of treatment for discoid lupus erythematosus. Dermatol Ther 2016; 29: 274.
Giacomel J, et al: Dermoscopy of hypertrophic lupus. J Am Acad Dermatol 2015; 72: s33.
Gulati G, Brunner HI: Environmental triggers in systemic lupus erythematosus. Semin Arthritis Rheum 2017 Oct 5; ePub ahead of print.
Jessop S, et al: Drugs for discoid lupus erythematosus. Cochrane Database Syst Rev 2017; 5: CD002954.
Khullar G, et al: Pigmented macular variant of chronic cutaneous lupus erythematosus. Clin Exp Dermatol 2017; 42: 793.
La Paglia GMC, et al: One year in review 2017: systemic lupus. Clin Exp Rheumatol 2017; 35: 551.
Liu RC, et al: Subacute cutaneous lupus erythematosus induced by nivolumab. Australas J Dermatol 2017 Jul 20; ePub ahead of print.
Maguiness SM, et al: Imiquimod-induced subacute cutaneous lupus erythematosus-like changes. Cutis 2015; 95: 349.
Mayor-Ibarguren A, et al: Subacute cutaneous lupus erythematosus induced by mitotane. JAMA Dermatol 2016; 152: 109.
Momen SE, et al: Tumour necrosis factor antagonist-induced lupus. Br J Dermatol 2017 Aug 3; ePub ahead of print.
Nieto-Rodriguez D, et al: Subacute cutaneous lupus erythematosus induced by masitinib. Int J Dermatol 2017; 56: 1180.
Nutan F, et al: Cutaneous lupus. J Investig Dermatol Symp Proc 2017; 18: s64.
Ramachandran SM, et al: Topical drug-induced subacute cutaneous lupus erythematosus isolated to the hands. Lupus Sci Med 2017; 4: e000207.
Ramezani M, et al: Diagnostic value of immunohistochemistry staining of Bcl-2, CD34, CD20 and CD3 for distinction between discoid lupus erythematosus and lichen planus. Indian J Pathol Microbiol 2017; 60: 172.
Rees F, et al: The worldwide incidence and prevalence of systemic lupus erythematosus. Rheumatology (Oxford) 2017; 56: 1945.
Romero-Mate A, et al: Successful treatment of recalcitrant discoid lupus erythematosus with ustekinumab. Dermatol Online J 2017; 23.
Shovman O, et al: Diverse patterns of anti-TNF-α-induced lupus. Clin Rheumatol 2017 Oct 23; ePub ahead of print.
Tiao J, et al: Using the American College of Rheumatology (ACR) and Systemic Lupus International Collaborating Clinics (SLICC) criteria to determine the diagnosis of systemic lupus erythematosus (SLE) in patients with subacute cutaneous lupus erythematosus (SCLE). J Am Acad Dermatol 2016; 74: 862.
Vincent JG, et al: Specificity of dermal mucin in the diagnosis of lupus erythematosus. J Cutan Pathol 2015; 42: 722.
Walsh NM, et al: Plasmacytoid dendritic cells in hypertrophic discoid lupus. J Cutan Pathol 2015; 42: 32.
Wieczorek IT, et al: Systemic symptoms in the progression of cutaneous to systemic lupus. JAMA Dermatol 2014; 150: 291.
Zuppa AA, et al: Neonatal lupus. Autoimmun Rev 2017; 16: 427.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


