Collagen-Vascular Diseases
Aaron Loyd MD
Gary Goldenberg MD
Joseph L. Jorizzo MD
The collagen vascular diseases are a group of autoimmune diseases including lupus erythematosus (LE), scleroderma, dermatomyositis (DM), rheumatoid arthritis, Sjögren’s syndrome, and many others. LE, scleroderma, and DM are discussed in detail herein. These diseases are characterized by autoimmune phenomena, including circulating autoantibodies (aAbs). There is a great deal of overlap of clinical and laboratory findings among these disorders. Cutaneous manifestations may be the presenting or dominant feature of collagent vascular disease. Nevertheless, systemic multiorgan disease needs to be excluded in each case.
A thorough laboratory evaluation is required. Much has been written about antinuclear antibody (ANA) testing. This assay identifies aAbs present in serum to autoantigens present in nuclei of mammalian cells. ANA testing is reported as a titer, and most commercial kits report ANA titers of 1:40 or 1:80 as abnormal. However, titers less than 1:160 are not diagnostic. Approximately 5% of otherwise healthy young individuals have an ANA titer of 1:160 or higher. The prevalence of this false-positive ANA increases with age. Therefore, it is important not to label patients with LE or another autoimmune disease simply based on one laboratory test. Additionally, ANA positivity is seen in multiple other autoimmune diseases and is not specific for LE.
After securing a diagnosis, much important work remains. A thorough medical history, including medicinal and social history, should be obtained. Paraneoplastic phenomena should also be considered, especially in the setting of DM. Finally, understanding the systemic effects of each disease is crucial for initial screening tests as well as appropriate referral.
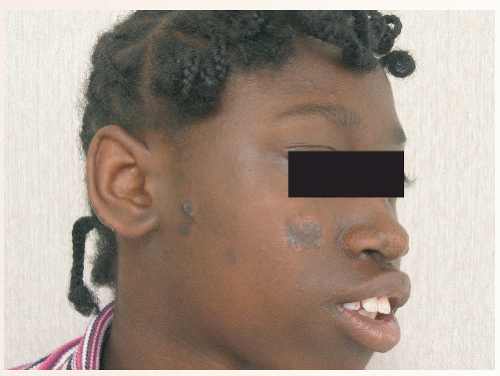 FIGURE 37-1 ▪ CCLE with discoid lesions—erythematous, hyperpigmented plaques with scarring characteristic of CCLE with discoid lesions. |
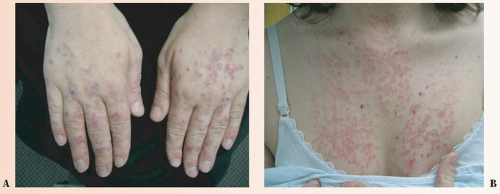 FIGURE 37-2 ▪ SCLE. (A) Erythematous papules and plaques on dorsal hands; typical distribution spares the knuckles. (B) Erythematous papules and plaques in a typical sun-exposed distribution of the V on the chest. |
Lupus Erythematosus
Chronic cutaneous lupus erythematosus (CCLE), subacute cutaneous lupus erythematosus (SCLE) (Figs. 37-1 and 37-2), and
acute cutaneous lupus erythematosus (ACLE) are the most common variants encountered by dermatologists.
acute cutaneous lupus erythematosus (ACLE) are the most common variants encountered by dermatologists.
SAUER’S NOTES
Smoking may decrease the benefit of antimalarials when used to treat LE and can cause a flare of cutaneous lupus. This is simply another in a list of seemingly endless reasons to stop smoking.
It has been estimated that cutaneous variants of LE are two to three times more common than systemic disease. LE is much more common in women than in men; the female to male ratio is at least 6:1. Young, fertile women are most commonly affected, and pregnancy issues should be addressed. LE is more common in blacks than in whites, and blacks have a higher frequency of systemic disease.
Drug-induced lupus has been reported with multiple medications, including hydrochlorothiazide, calcium channel blockers, isoniazid, phenytoin, angiotensin-converting enzyme inhibitors (captopril), tetracyclines (minocycline), and many others including over-the-counter nonsteroidal anti-inflammatory drugs. Antihistone aAbs is a serologic marker that occurs in patients with drug-induced LE.
Neonatal LE (NLE) is seen in newborns whose mothers have anti-Ro aAbs. Nearly 100% of patients with NLE also have anti-Ro aAbs. SCLE-like lesions are usually seen on the face of newborns. Congenital heart block is an important complication and is usually present at birth. Therefore, screening for heart disease should be performed in all patients with NLE. Children with this complication have a 20% mortality rate and two thirds require pacemakers.
Diagnosis of LE and its subtypes depends on a constellation of findings that include history, physical examination, histopathologic correlation, and laboratory evaluation that focuses on published American College of Rheumatology criteria (Table 37-1). See Table 37-2 for a comparison of CCLE, SCLE, and ACLE. The treatment approach is outlined in Table 37-3.
Scleroderma
Localized scleroderma (morphea), systemic (diffuse) sclerosis, and CREST syndrome (calcinosis cutis, Raynaud’s phenomenon, esophageal dysfunction, sclerodactyly, telangiectasias) are variants included under the general heading of scleroderma (Fig. 37-3).
Systemic sclerosis (SSc) is a systemic disease that affects the skin, lungs, heart, gastrointestinal (GI) tract, and other organ systems. It is upto 15 times more common in women, and the age of onset is between 30 and 50 years. Ten-year survival of patients with SSc is under 70%. Pathogenesis of SSc is unknown, but endothelial cell damage is suspected as the key pathogenic abnormality.
aAbs are often detected in nonlocalized forms of sclerosis. ANA may be used as a screen, while Scl 70 antibodies differentiate SSc from the centromere antibody-associated CREST syndrome. Cutaneous and systemic findings of SSc and other variants of scleroderma are summarized in Table 37-4.
TABLE 37-1 ▪ Systemic Lupus Erythematosus Criteria (4 of 11 needed for diagnosis of systemic disease) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
Dermatomyositis
DM is an autoimmune proximal extensor inflammatory myopathy with specific cutaneous manifestations (Fig. 37-4). Polymyositis is an inflammatory myopathy without any skin involvement. DM sine myositis presents with the characteristic skin eruption of DM without muscle involvement.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


