Key points
- •
Less than 50% of patients affected with atopic dermatitis have a genetic mutation of the skin barrier protein, filaggrin. Thus additional contributing factors, barrier defects, and beyond could account for disease triggering of the other 50% of patients with atopic dermatitis but without filaggrin mutation.
- •
Patients affected with ichthyosis vulgaris and genetic defect of skin barrier protein filaggrin do not all develop atopic dermatitis. Thus skin barrier defect-enabled external triggers are not the only factors contributing to the atopic dermatitis development.
- •
Patients with atopic dermatitis have an abnormal “resting” internal immune milieu during disease remission, suggesting an abnormal internal immune milieu as a contributing factor. Additionally, patients with atopic dermatitis have other comorbidities suggesting an underlying immune abnormality. Moreover, the atopic march phenomenon may indicate an internal immune milieu that allows such “atopic expansion.”
- •
Patients with a history atopic dermatitis can suffer eczema vaccinatum, a life-threatening, widespread vaccinia virus with systemic illness when exposed to smallpox vaccine, even at a time of clinical inactivity of atopic dermatitis, providing further support of an internal triggering factor.
- •
Patients with atopic dermatitis history can suffer eczema herpeticum, a widespread herpetic infection when exposed to herpes simplex virus, even at a time of clinical inactivity of atopic dermatitis, again supporting an abnormal internal milieu.
Introduction
With delineated evidence gathered from laboratory studies at the immunologic and molecular levels to implicate external contributing factors of atopic dermatitis (see Chapter 11, Chapter 12, Chapter 13, Chapter 14, Chapter 15, Chapter 16, Chapter 17 ), we now examine the internal factors from the perspective of clinical evidence. Although clinical evidence is not as robust as that collected through the immunologic or molecular level of investigation, it nevertheless provides a supporting documentation from a unique angle. Clinical evidence provides a sense of reality, a sense of living proof, so to speak. In fact, any medical theory or purely laboratory-based research result unsupported by clinical evidence will be deemed invalid for the actual disease.
The Oxford Living Dictionaries defines the term as “relating to the observation and treatment of actual patients rather than theoretical or laboratory studies… (of a disease or condition) causing observable and recognizable symptoms.” The term is defined in Merriam-Webster ( ) as “of, relating to, or conducted in or as if in a clinic: such as a: involving direct observation of the patient or b: based on or characterized by observable and diagnosable symptoms.” Clinical evidence therefore is data gathered from the clinical observations or clinical studies.
To collect clinical evidence, we gather all the data relating to the symptoms and signs of what we can observe, obtain, and measure about and from actual patient encounters, rather than by theoretic consideration, speculation, or purely laboratory investigation. Nevertheless, laboratory data are also part of the supporting evaluation of clinical data and of documenting clinical evidence. One simple example is the clinical evaluation of early clinical failure of treatment of a gram-negative bacteria sepsis (bloodstream infection). To collect clinical evidence accurately and correctly, the clinical investigators must first establish that all these patients indeed have bloodstream infection by gram-negative bacteria documented by results of blood culture, a laboratory method. In addition, the clinical researchers must measure many parameters to develop a set of criteria for determining the predictors for early clinical failure. These parameters would include “purely clinical data” such as blood pressure, respiratory rate, altered mental status, but they would also include “laboratory data” such as white blood cell count. Together, these data provide the valuable predictors for early clinical failure on treatment for gram-negative sepsis ( ). Similarly, to collect clinical data and to document clinical evidence in relation to development of atopic dermatitis, some laboratory-generated information, such as serum level of immunoglobulin E (IgE), bacterial culture results, skin histopathology report, genetic mutation information in skin barrier protein, immunologic status, and other clinically supportive laboratory data, is also included. The following discussions delineate some reasons to suggest that internal factors play a significant role in atopic dermatitis development.
Skin barrier defect is not synonymous with atopic dermatitis
One of the reasonable arguments against external factor as the sole contributor for atopic dermatitis goes like this: If the cause of atopic dermatitis is exclusively external, then it should naturally follow that in those patients with skin barrier defect, the resulting easy entry of the offending substance (pathogens or allergens) into the skin should, eventually, lead to development of chronic immune reactions in all patients. Therefore we should find that all patients with skin barrier defect should eventually develop atopic dermatitis when they reach adulthood. Is this conclusion supported by clinical evidence? The answer is a definite no. Outside-in dysregulation leads to atopic dermatitis, but apparently not in all cases.
Not all patients with filaggrin gene mutation develop atopic dermatitis
Filaggrin, one of the major skin barrier proteins that form a protective layer at the stratum corneum level of the skin, is now well documented to be defective, due to genetic mutation, in patients affected by an ichthyosis condition termed ichthyosis vulgaris, with a documented reduction in thickness of granular layer of the epidermis under electron microscopy ( ; ). In addition, filaggrin mutation in ichthyosis vulgaris causes various abnormal structure organization in the epidermis, including perinuclear retraction of keratin filaments, impaired loading of lamellar body contents, nonuniform extracellular distribution of secreted organelle contents, and abnormal lamellar bilayer architecture. These structural abnormalities predictably link to the functional defect, such as increased transepidermal water loss, an objective indicator of defective skin barrier function ( ). If an external factor is the only component needed to induce atopic dermatitis, one would expect that sooner or later all patients affected by ichthyosis vulgaris will develop atopic dermatitis, as our reason goes. However, this does not seem to be the clinical finding. First, it is now clear that not all patients affected with ichthyosis vulgaris develop atopic dermatitis, and this fact is well documented by reports published in different parts of the world ( ; ; ; ). A study showed that even with compound heterozygosity (biallelic filaggrin null mutation), some patients affected by ichthyosis vulgaris do not develop the atopic dermatitis disease ( ). In a study report published in 2009, there is a significantly higher number of antigen presenting cells (CD1a+ Langerhans cells) present in the nonlesional epidermis of ichthyosis vulgaris patients affected with atopic dermatitis than those patients unaffected with atopic dermatitis. Thus the finding of this study suggests that an immunologic factor other than or in addition to epidermal barrier defect may account for the atopic dermatitis development ( ).
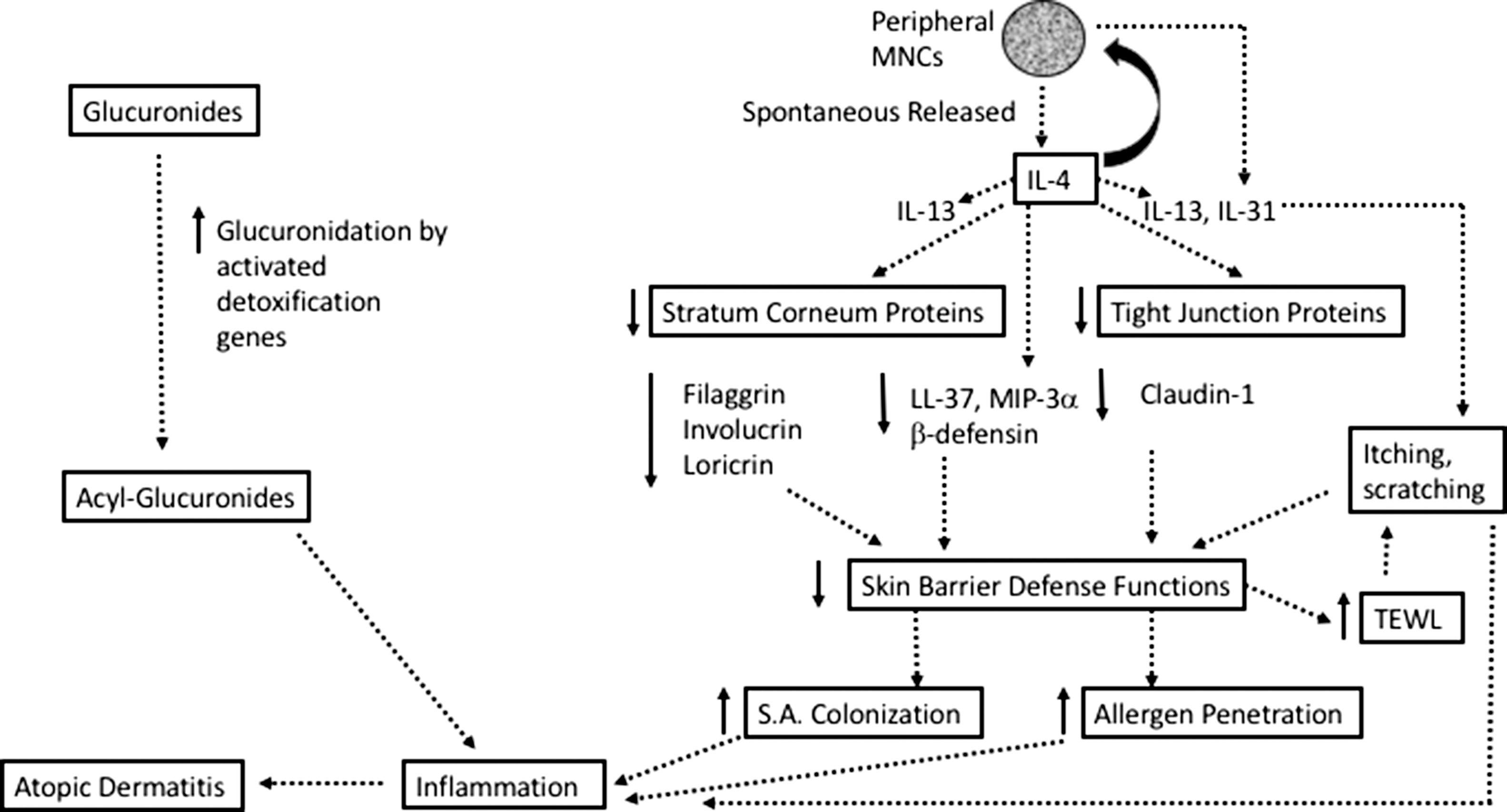
Another recently reported finding of enhanced expression of genes related to xenobiotic metabolism occurred only in the nonlesional skin of patients with atopic dermatitis, not in those patients with ichthyosis vulgaris, which also supports the notion that factors other than skin barrier defect of filaggrin may be involved in the development of atopic dermatitis. Specifically, investigators found that the genes involved in the processing of pollutants, endocrine disruptors, and xenobiotics, particularly glucuronidation, were substantially upregulated in the skin of atopic dermatitis patients but not in that of ichthyosis vulgaris patients. Glucuronidation is a chemical process that converts the less biologically active form of glucuronides to reactive metabolite acylglucuronides ( ). In other words, detoxification genes are highly activated in atopic dermatitis. Importantly, these enhancements exhibited in the skin of patients affected with atopic dermatitis, with or without filaggrin gene mutation, further reduce the importance of filaggrin gene mutation as a sole triggering factor for atopic dermatitis. These investigators suggested that an inflammatory triggering of local metabolism of noxious molecules might be the key factor that could transform a subinflammatory skin to an overt inflamed skin observed in atopic dermatitis ( ).
Not all atopic dermatitis patients have an identifiable filaggrin gene mutation
Conversely, we can ask the question from the opposite direction: Do all patients affected by atopic dermatitis have the mutation of major skin barrier protein filaggrin? The answer is, again, no ( ; ; ; ; ; ; ; ). An early study based in Japan indicates that only a little over 20% of atopic dermatitis patients carry some form of filaggrin gene mutations ( ). A subsequent study in Japanese atopic dermatitis patients reveals that just 25% of patients carry one or more filaggrin mutations ( ), significantly lower than the high percentage (near 50%) of mutation present in European atopic dermatitis patients ( ; ). Similarly, in a study of Singaporean patients with atopic dermatitis, only 20% carry one or more filaggrin gene mutations ( ). Further, filaggrin mutations occur in Han Chinese atopic dermatitis patients with an overall rate of 31% ( ; ). Among African patients affected by both atopic dermatitis and ichthyosis vulgaris, a study documents that only 22% of these patients carry heterozygous filaggrin mutation ( ). In a study of Korean patients with atopic dermatitis, the investigators discovered that only 68 among the 1110 atopic dermatitis patients have filaggrin mutations (6.1%): 18 patients have the heterozygous 3321 delA mutations, 49 patients have the heterozygous, and 1 patient has the homozygous p.K4022X mutations ( ). Another study of Korean atopic dermatitis showed that four filaggrin null mutations (3321 delA, K4022X, S3296X, and S2889X) were detected in only 16% of the 70 tested patients ( ).
Not surprisingly, patients with filaggrin mutations tend to have poorer skin hydration, earlier disease onset, and more severe atopic dermatitis ( ). Overall, approximately 10% to 50% of patients in different cohorts affected by atopic dermatitis harbor mutations at different regions of the filaggrin gene ( ; ). The next logical question to ask is: Do those atopic dermatitis patients who have no filaggrin gene mutation have genetic mutations of other skin barrier proteins such as loricrin or involucrin? Barrier dysfunctions due to yet-to-be-discovered mutations of those nonfilaggrin barrier proteins, if existed, may account for the external triggering factor in atopic dermatitis. At the present time, however, we do not have the answer, and there is no literature report as of June 2020. This question is important to answer as it would help decipher whether atopic dermatitis is a pure inside-out or outside-in process, or a combination of both. The following paragraphs will examine the internal immune milieu of patients affected by atopic dermatitis.
Th2 immune type correlates with inside-out dysregulation in atopic dermatitis
The internal “resting” immune milieu is skewed toward Th2 in atopic dermatitis patients
Another reasoning argues against external factor as the sole contributor for atopic dermatitis is as follows: If the cause of atopic dermatitis is exclusively external, then the immunologic status of the patients affected by atopic dermatitis should remain normal when the patient is lesion free and would become activated only when it is in a lesional condition. The best way to answer this question is to examine the immune milieu of these patients when their skin disease is inactive. The data obtained during inactive disease stage are essential for determining the internal immune milieu because once the disease is active the immune data may simply reflect a reactive immune state rather than a resting immune state. Study results, although somewhat limited thus far, point to an abnormal resting immune system in atopic dermatitis patients ( ; ; ; ; ).
Abnormal upregulation of Th2-type cytokines
The Th2-type immune milieu is essential for the development of any allergic diseases. IL4, a key Th2 cytokine, is critical for IgE production and is the driver for Th2 lymphocyte development and atopic diseases ( ; ; ; ). IL13 is a Th2 cytokine with its gene located close to IL4 gene on the same chromosome, 5q31, and its protein shares the same cellular receptor (IL4 receptor-α) with IL4. Both IL4 and IL13 are known for their stimulatory role in allergic diseases ( ; ). Studies have also shown a higher level of IL13 than IL4 in atopic dermatitis skin lesions ( ). Besides IL4 and IL13, evidence shows that IL10 plays a significant role in allergic diseases ( ).
Animal models further support the role of the Th2 immune milieu in atopic dermatitis. Epidermally expressed IL4 by transgenesis in mice induces a chronic, pruritic, inflammatory skin disease that fulfills the clinical diagnosis of human atopic dermatitis. Before the disease onset, there are already upregulations of Th2-type cytokines (IL4, IL5, IL6, IL13) detected in the “normal appearing” skin of these IL4-transgenic mice, and the Th2 cytokine upregulation went higher still after the onset of skin lesions ( ; ). Epidermal expression of IL4 in BALB/c mice, a strain with principal internal Th2 immune milieu, induces a much earlier disease onset and a more severe skin inflammation than IL4 expressing in C57BL/6 mice, a strain with predominant internal Th1 immune milieu ( ; ).
Patients with atopic dermatitis have an immune dysregulation and a predominant Th2-type cytokine milieu ( ). The Th2 immune milieu is also suppressive to immune defense against certain infectious microorganisms. One of the best clinical examples is observed in the disease of leprosy, a chronic bacterial infection caused by Mycobacterium leprae . Patients with a milder/resistant form of tuberculous leprosy have strong skin mRNA expressions for IFN-γ and IL2, the Th1-type cytokines; patients with a severe/multibacillary form of lepromatous leprosy, however, have skin cytokine mRNAs predominant for IL4, IL5, and IL10, the Th2 types ( ). Human allergen-specific CD4+ T-cell clones obtained from atopic patients produce Th2 cytokines IL4, and not Th1 cytokine IFN-γ, when stimulated in the presence of autologous antigen presenting cells, regardless if the antigen presenting cells are derived from active or inactive disease state. By contrast, T-cell clones obtained from nonatopic donors exposed to the same allergen produce Th1 cytokine IFN-γ under the identical stimulating condition, further supporting the Th2-skewed immune milieu in the resting state of atopic dermatitis ( ).
Peripheral blood mononuclear cells derived from atopic dermatitis patients have a greater tendency for spontaneous release of IL4 and IL10, suggesting T cells of atopic dermatitis are skewed toward a Th2 response ( ; ). Secretion of IL4 induces expression and secretion of the pruritus-inducing cytokine IL31 from helper T cells and mast cells, further amplifying Th2 cytokine effects in atopic diseases ( ), and leads to constant itch-scratch cycles exacerbating the chronic skin inflammation ( ).
In the absence of a comprehensive study on the nonlesional skin cytokine milieu or skin cytokine milieu during the inactive disease stage of atopic patients, one can still learn some useful insights by comparing the cytokine milieu between two common inflammatory skin diseases, atopic dermatitis and psoriasis. In contrast to the prominent Th2 immune milieu in atopic dermatitis skin, psoriasis skin is marked with a strong Th1 immune response. Evidence obtained from lesional skin of atopic dermatitis patients shows an inability to produce sufficient amounts of human defensins and inflammatory cytokines tumor necrosis factor-α (TNF-α) and IFN-γ ( ). Moreover, Th2 cytokines, IL13 and IL4, inhibit TNF-α- and IFN-γ-induced human β-defensin production ( ). These findings help define the cytokine patterns that drive immune deviation toward a Th2 immune response in atopic dermatitis, in contrast to that of psoriasis.
Atopic dermatitis patients have immune-mediated comorbidities
Besides the documented Th2 immune deviations, some patients affected by atopic dermatitis also have comorbidities related to underlying immunologic conditions. These include autoimmune diseases such as alopecia areata, gastrointestinal immune-mediated disorders, and cardiovascular diseases ( ; ). The occurrence of these comorbidities suggests an underlying immune deviation in patients affected by atopic dermatitis. However, it is not completely clear if these comorbidities exist before atopic dermatitis develops or they occur as a sequela of atopic dermatitis development.
Th2 immune response cytokines suppress expressions of skin barrier proteins
While we would like to highlight the distinct properties of outside-in and inside-out dysregulation in atopic dermatitis, the two are intimately connected. Study results show that the major Th2 cytokines, IL4 and IL13, alone or in combination could suppress the expression of three critical stratum corneum–located primary skin barrier proteins: filaggrin, involucrin, and loricrin ( ; ). In addition, the combination of three Th2 cytokines (IL4, IL13, and IL31) suppresses the expression of claudin-1, a tight junction protein important for skin barrier function ( ). These data show that internal immune derangements observed in atopic dermatitis could induce skin barrier defects even in the absence of genetic mutations. Thus internal immune deviation (an inside-out dysregulation) could lead to external contributing factors: skin barrier weakness–related penetration of pathogens and allergens (an outside-in dysregulation), provoking the development of atopic dermatitis. More details on this aspect are discussed in Chapter 11 .
Atopic March
One of the well-known clinical phenomena in relationship to atopic dermatitis is atopic march, which documents the occurrence of extracutaneous atopic diseases such as asthma, food allergy, and allergic rhinitis after patients developed cutaneous atopic dermatitis in early childhood ( ). While the pathomechanism of atopic march is not completely understood and is being pursued by physicians and scientists in the field, there is some evidence pointing to an internal immune dysregulation. For example, the majority of IgE detected from patients with asthma is not accounted for by known allergens in patients with highest total IgE levels, supporting a notion of internal IgE dysregulation. A history of infant-onset atopic dermatitis together with a parental family history of atopy increased the risk of subsequent upper or lower airway allergic manifestations, suggesting a genetic link of atopy. Accordingly, field experts support recommendations to examine if early immune interventions utilizing systemic Th2 inhibition could prevent atopic march in high-risk children with atopic dermatitis ( ).
Increased risk of atopic dermatitis–associated viral infections
Observations of increased viral-related skin infections in patients of atopic dermatitis include eczema vaccinatum, eczema herpeticum, eczema coxsackium, and a higher occurrence of human papillomavirus infections. The details of these viral infections are delineated next.
Eczema vaccinatum
The most dramatic clinical evidence illustrating an abnormal immune milieu in atopic dermatitis is none other than the rare occurrence of eczema vaccinatum. In such event, patients with atopic dermatitis develop systemic illness and widespread smallpox lesions after being exposed to people recently vaccinated against smallpox. Eczema vaccinatum can be fatal and can occur in patients with or without active dermatitis lesions, even in patients who have had no active skin disease for years. Results of early and recent studies suggest this phenomenon occurs because of altered immune defense against the smallpox viruses ( ; ; ; ; ).
Investigators have demonstrated an enhanced vaccinia virus replication in skin samples from nonlesional skin of atopic dermatitis patients, compared to skin samples from psoriasis patients and healthy individuals in an ex vivo experiment and linked to LL-37. LL-37 is an antimicrobial peptide known to suppress vaccinia virus replication in a dose-dependent manner. In nonlesional skin of atopic dermatitis patients, vaccinia viruses fail to induce expression of LL-37, in contrast to LL-37 induction observed in nonlesional skin from psoriasis patients and normal individuals. Furthermore, the combination of Th2 cytokines IL4 and IL13 enhances the vaccinia virus replication and suppresses expression of LL-37 by the vaccinia virus–stimulated keratinocytes ( ). In the clinical situation, increased levels of IL4 and IL13 in atopic dermatitis would likely suppress LL-37 expression, thereby contributing to unchecked replication of vaccinia viruses as observed in eczema vaccinatum.
Additional evidence pointing to pathomechanism of eczema vaccinatum is from the study of macrophage inflammatory protein 3 alpha (MIP-3α), a C-C chemokine that plays an important role in innate and adaptive immune response and has documented antimicrobial activities against the vaccinia virus. Investigators have demonstrated that the skin lesions from atopic dermatitis patients exhibited only 50% of the MIP-3α expressed by that of psoriasis patients. Furthermore, preincubation with IL4 and IL13 significantly suppresses the induced expression of this chemokine in keratinocytes when stimulated with the vaccinia virus ( ).
Eczema herpeticum
Another excellent clinical evidence to support an altered resting immune milieu in patients affected by atopic dermatitis is the clinical disease of eczema herpeticum. Eczema herpeticum, also termed Kaposi varicelliform eruption, occurs when atopic dermatitis patients develop widespread herpetic blistering eruptions secondary to exposure to herpes simplex viruses ( ; ). Eczema herpeticum manifests with fever, viremia, skin erosion, lymphadenopathy, keratoconjunctivitis, and (in severe cases) meningitis. The fact that it occurs in patients with or without active skin disease further supports the notion of an altered resting immune milieu in atopic dermatitis patients ( ).
In addition, these atopic dermatitis patients have a significantly higher level of total serum IgE ( ; ; ), and there is an inverse correlation between IgE level and LL-37 expression in the skin ( ). Patients with a history of eczema herpeticum have a defect in their skin’s ability to induce expressions of HBD-2, HBD-3, and cathelicidin (LL-37), the three important epidermis-produced antimicrobial peptides. They also have a higher level of Th2 cytokine IL13 in nonlesional skin, compared to those atopic dermatitis patients with no history of eczema herpeticum. This high level of IL13 may predispose these patients for eczema herpeticum development ( ; ).
The tendency to develop eczema herpeticum has been linked with STAT6 single nucleotide polymorphisms (SNPs), particularly at a 2-SNP (CT) haplotype ( ). Resting peripheral blood CD8+ T cells obtained from atopic dermatitis patients with a history of eczema herpeticum have defective interferon-gamma (IFN-γ) production in response to stimulation with herpes simplex virus in vitro, and these patients have an HLA-B7 genotype ( ). Moreover, IFN-α and IL29 mRNAs and proteins are significantly reduced in the atopic dermatitis patients with a history of eczema herpeticum, compared to those patients without the history of eczema herpeticum, when their resting peripheral blood mononuclear cells are stimulated with herpes virus in culture. Further studies document a significant inhibition of upstream regulators IRF3, IRF7, and IRF9 in atopic patients with the history of eczema herpeticum. IRFs are important transcription factors involving in a good deal of viral recognition signaling pathways, and some examples are toll-like receptors, cytoplasmic DNA sensors, and cytoplasmic RNA sensor RIG1 ( ). Subsequently, atopic dermatitis patients with the history of eczema herpeticum have shown functional defect in genetic variants of the interferon-pathway gene encoding interferon-gamma receptor 1 (IFNGR1) ( ).
Eczema coxsackium
Atopic dermatitis patients have also developed eczema coxsackium, extensive skin blistering, when exposed to Coxsackie viruses, and it can occur in the absence of active atopic dermatitis skin lesions. In contrast to the limited skin involvement observed in the typical Coxsackie viral infection (i.e., hand, foot, and mouth disease), eczema coxsackium manifests with fever and widespread lesions of papules, blisters, and erosions, a clinical morphology similar to that of eczema herpeticum ( ; ). Although the pathophysiology of eczema coxsackium is not fully understood, an altered internal immune response may play an important role, as documented in the study results on eczema herpeticum and eczema vaccinatum ( ; ; ; ).
High-risk cervical human papillomavirus infection
Clinical evidence demonstrates an association between increased infections of high-risk cervical human papillomavirus and female atopic dermatitis patients ( ). Atopic dermatitis is significantly more commonly identified in patients affected with high-risk human papillomavirus infection (8.3%) than in patients with negative infection ( P = .007). Since high-risk human papillomavirus infection is more likely to persist in immunosuppressed women, the data seem to suggest that atopic dermatitis is associated with a level of immunosuppression or an altered immune response as discussed earlier.
Summary
We have examined available clinical evidence in relation to the development of atopic dermatitis to pursue an answer whether internal or external contributing factors are triggers for atopic dermatitis development. From the data showing that not all patients with ichthyosis vulgaris and skin barrier protein filaggrin mutation developed atopic dermatitis, that not all patients affected by atopic dermatitis have identified filaggrin mutation, that some internal immune milieu of Th2 deviations existed in patients with atopic dermatitis, that Th2 cytokines can suppress skin barrier protein expression in the absence of gene mutation, and that widespread viral infection (such as eczema vaccinatum, eczema herpeticum, and eczema coxsackium) can occur in patients with a history of atopic dermatitis but no active skin disease, the hypothesis that all cases of atopic dermatitis can be developed without an altered internal immune milieu cannot be supported. Fig. 18.1 depicts a proposed mechanism in which internal immune deviation toward Th2 milieu would trigger the type of inflammation observed in atopic dermatitis.